Лекарственные средства, действующие преимущественно на центральную нервную систему [11]
Ондансетрон (эметрон, зофран)
Тиэтилперазин (торекан, горестен, трестен)
Дифенгидрамин (димедрол)
Синтез аминазина
Синтез анестезина
Синтез анестезина из р-толуидина
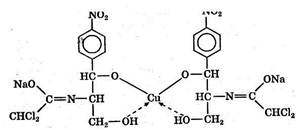
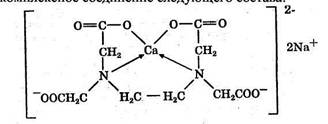
Синтез валидола
Мл 0,1 н. раствора хлорной кислоты соответствует 0,03038 г C17H17NO2 * HCl, которого в пересчете на сухое вещество должно быть не менее 98,0%
Мл 0,1 мол раствора нитрита натрия соответствует 0,01652 г С9Н11NО2, которого в препарате должно быть не менее 99,5 %
Навигация
Дифенгидрамин (димедрол)
Рвотные и противорвотные препараты
131931
знак
9
таблиц
27
изображений
4.3.1 Дифенгидрамин (димедрол).
Является одним из основных представителей группы противогистаминных препаратов, блокирующих Н1-рецепторы [11]. Он обладает весьма выраженной противогистаминной активностью. Кроме того, он оказывает местноанестезирующее действие, расслабляет гладкую мускулатуру в результате непосредственного спазмолитического действия, блокирует в умеренной степени холинорецепторы вегетативных нервных узлов.
Димедрол хорошо всасывается при приеме внутрь. Проникает через гематоэнцефалический барьер.
Важной особенностью димедрола является его седативное действие, имеющее некоторое сходство с действием нейролептических веществ; в соответствующих дозах он оказывает снотворный эффект. Является также умеренным противорвотным средством. В действии димедрола на нервную систему существенное значение имеет наряду с влиянием на гистаминовые рецепторы (возможно, Н3 – рецепторы мозга) его центральная холинолитическая активность.
4.3.2 Прометазин (дипразин, фенерган).
Обладает сильной противогистаминной активностью (более активен, чем димедрол) [13]. Дипразин является производным фенотиазина; по строению, а частично и по фармакологическим свойствам близок к аминазину. Наиболее важной фармакологической особенностью дипразина является его сильная противогистаминная (Н1-блокирующая) активность
Дипразин хорошо всасывается при приеме внутрь. При разных путях введения проникает через гематоэнцефалический барьер.
Препарат оказывает выраженное влияние на ЦНС; обладает довольно сильной седативной активностью, усиливает действие наркотических, снотворных, аналгезирующих и местноанестезирующих средств, понижает температуру тела, предупреждает и успокаивает рвоту. Он оказывает также умеренное периферическое и центральное холинолитическое действие. Сильно выражено адренолитическое действие дипразина.
4.4 М-холиноблокаторы
4.4.1 Скополамин.
Химически скополамин близок к атропину: является сложным эфиром скопина и троповой кислоты [11]. Близок к атропину по влиянию на периферические холинореактивные системы. Подобно атропину вызывает расширение зрачков, паралич аккомодации, учащение сердечных сокращений, расслабление гладких мышц, уменьшение секреции пищеварительных и потовых желез
Оказывает также центральное холинолитическое действие. Обычно вызывает седативный эффект: уменьшает двигательную активность, может оказать снотворное действие. Характерным свойством скополамина является вызываемая им амнезия.
5. Методы получения РВОТНЫХ И ПРОТИВОРВОТНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ 5.1 Синтез рвотных лекарственных средств 5.1.1 Синтез апоморфинаХимический процесс [14]:
![]()
морфин апоморфин
Получение: 1 ч. чистого морфина и 10 ч. 25%-ной соляной кислоты нагревают в запаянной трубке в течение 2–3 часов при 140–150 °С. По охлаждении к содержимому трубки прибавляют избыток двууглекислого натрия и жидкость быстро взбалтывают (при возможном отсутствии воздуха) с эфиром или хлороформом. При этом неизменившийся морфин остается нерастворенным. К раствору апоморфина в эфире или хлороформе приливают небольшое количество крепкой соляной кислоты и выделившуюся хлористоводородную соль перекристаллизовывают из небольшого количества горячей воды. Из очищенной таким образом хлористоводородной соли выделяют свободное основание, прибавив к раствору соли двууглекислой соды.
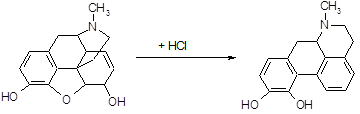
Апоморфин получается при нагревании морфина с 25 %-ной соляной кислотой в автоклаве при температуре 130–140°С в течение 2–3 часов. При этом от морфина отщепляется молекула воды [15]:
![]()
Под действием кислот разрывается кислородный мостик и этаминная цепь перемещается из положения 13 в положение 8. Происходит перегруппировка с превращением морфина в апоморфин (левовращающий). Его химическое строение отличается от строения морфина: апоморфин представляет собой почти плоскую молекулу, которую можно рассматривать как производное фенантрена и изохинолина.
Так как апоморфин-основание крайне нестоек, его применяют в виде хлористводородной соли. Поэтому полученную массу нейтрализуют содой и извлекают эфиром. К эфирной вытяжке добавляют раствор соляной кислоты и тщательно перемешивают; выделяется белый осадок хлористводородного апоморфина.
Апоморфин в малых дозах угнетает активность дофаминергической системы и вызывает седативный эффект у животных [16]. Имеются данные об использовании апоморфина для лечения психотических нарушений при алкогольном абстинентном синдроме и шизофрении, когда наблюдается повышение активности дофаминергической системы. Однако наличие нежелательных побочных эффектов, как высокая эметическая активность и кратковременность действия, осложняют его применение в клинике. В связи с этим были проведены синтез и фармакологическое изучение некоторых О, О´-диацилпроизводных апоморфина (I – VI) с целью изыскания соединений, лишенных указанных недостатков.

I: R = COC6H4-Br-4; II: R = COC6H4-F-4; III: R = COC6H4-CH3-4; IV: COC6H4-OPr-4; V: R = Ac; VI: R = COPh.
Экспериментальная химическая часть
ИК-спектры соединений регистрировали на спектрометре PE-580 (США) в вазелиновом масле, спектры ПМР получены на приборе «Varian» (60 МГц), внутренний стандарт – ГМДС.
О, О´-ди(4-бромбензоил)апоморфин (I). К раствору 1 г (3,2 ммоля) гидрохлорида апоморфина в смеси 3 мл диглима и 5 мл абсолютного пиридина прибавляют по каплям в токе азота 2,1 г (9,6 ммоля) хлорангидрида 4-бромбензойной кислоты. Реакционную смесь нагревают в течение 1 ч при 100°С в токе азота, выливают в 25 мл ледяной воды и эктрагируют CHCl3. Экстракт промывают насыщенным раствором NaHCO3 и NaCl, сушат безводным MgSO4 и упаривают. Получают 1,74 г I. Выходы, константы и данные спектров полученных веществ приведены в табл. 1.
Таблица 5.1
Производные апоморфина (I –IV)
| Соединение | Выход, % | Т.пл., °С | Найдено, % | Брутто- формула | Вычислено, % | ИК-спектр, υСО, см-1 | ||||
| С | Н | N | С | Н | N | |||||
| I | 86 | 197-8 | 58,73 | 3,78 | … | C31H232NO4 | 58,78 | 3,76 | … | 1750 |
| II | 75 | 194-6 | … | … | 2,85 | C31H23F2NO4 | … | … | 2,73 | 1750 |
| III | 81 | 184-5 | 79,02 | 5,82 | 3,13 | C33H27NO4 | 78,69 | 5,80 | 2,79 | |
| IV | 40 | 210-2,5 | 68,38 | 5,89 | 2,30 | C37H37NO6 * * 0,5C10H8O6S2 | 68,55 | 5,61 | 1,96 | |
Примечание. Соединение V, т.пл. 124°С и VI, т.пл. 157°С. Для I найдено, %: Br 25,62. Вычислено, %: Br 25,24. Соединения I, II, IV очищены перекристаллизацией из спирта, соединение III – из CHCl3. Соотношение апоморфинового и нафталиндисульфокислотного компонентов (2:1) у соли IV подтверждено спектром ПМР.
О,О´-ди(4-фторбензоил)апоморфин (II). Из 1 г (3,2 ммоля) гидрохлорида апоморфина и 1,5 г (9,6 ммоля) хлорангидрида 4-фторбензойной кислоты аналогично соединению I получают 1,37 II.
Геми-нафталин-1,5-дисульфонат О,О´-ди(4-пропоксибензоил)апоморфина. Аналогично из 1 г (3,2 ммоля) гидрохлорида апоморфина и 1,9 г (9,6 ммоля) хлорангидрида 4-пропоксибензойной кислоты с последующей обработкой основания IV раствором нафталин-1,5-дисульфокислоты в спирте получают 0,94 г геми-нафталин-1,5-дисульфонат IV.
О,О´-ди(4-метилбензоил)апоморфин (III). Смесь 1 г (3,2 ммоля) гидрохлорида апоморфина и 5 г (32 ммоля) 4-толуиловой кислоты в 10 мл CF3COOH нагревают в течение 1 ч при 100–110°С. Реакционную смесь упаривают в вакууме, остаток встряхивают с 50 мл эфира и 30 мл насыщенного раствора NaHCO3. Эфирный раствор отделяют, промывают водой, упаривают и получают 1,04 г III.
5.1.3 Синтез бромокриптинаПромышленные методы получения алкалоидов спорыньи, применяемые за рубежом, основаны на извлечении их из предварительно обезжиренной спорыньи органическими растворителями [17].
В Харьковском научно-исследовательском химико-фармацевтическом институте предложен метод избирательной водной зкстракции алкалоидов из спорыньи, в результате которой отдельно получают зкстракты, содержащие эргометрин, и экстракты, содержащие полипептидные алкалоиды. Первые используют для получения из них зргометрина, вторые – для выделения эрготоксина и эрготамина.
Экстракты, содержащие алкалоиды полипептидного типа, прозрачны, слабо окрашены; алкалоидов в них содержится 0,2–0,4 мг/мл (в зависимости от содержания их в исходной спорынье), экстрактивных веществ – 0,2–0,3 %, рН экстрактов – около 2,0.
Таблица 5.2
Алкалоидный состав ряда образцов ржаной спорыньи
| Место сбора спорыньи | Общее содержание алкалоидов (в % к весу спорыньи) | Группа | Правовращающие группы эрготоксина | |||
| эрготамина | эрготоксина | |||||
| эрготамин | эрготаминин | эргокристин + эргокорнин | эргокриптин | |||
| в % к общей сумме алкалоидов | ||||||
| Киевская область | 0,140 | 17,5 | Следы | 36,3 | 17,5 | 11,7 |
| » » | 0,266 | 20,0 | 0 | 60,6 | 0 | 0 |
| Харьковская область | 0,300 | 16,8 | Следы | 49,1 | Следы | 3,6 |
Примечание. Все данные приведены в пересчете на эргокристин. Общее содержание алкалоидов определяли по водному методу, отдельные алкалоиды – методом хроматографии на бумаге.
Алкалоидный состав зкстрактов устанавливали методом хроматографии на бумаге в системе бензол – формамид. Он соответствовал составу исходной спорыньи. Исходной спорыньей служили разные образцы дикорастущей ржаной спорьньи. В таблице приведен алкалоидный состав некоторых наиболее характерных образцов.
Алкалоиды выделяли из экстрактов по схеме получения эрготала. Алкалоиды высаливали добавлением 20–25 % раствора хлористого натрия. Из выпавшего осадка их извлекали хлороформом в щелочной среде и после концентрирования хлороформных экстрактов осаждали петролейным эфором. Выделенный при этом продукт представлял собой смесь нерастворимых в воде алкалоидов с [a]D20 = –20°, –60° (с 1, хлороформ). Эту смесь обрабатывали фосфорной кислотой в ацетоновом растворе с целью перевода ее полностью в левовращающие, физиологически активные алкалоиды. Полученные фосфорнокислые соли алкалоидов переводили затем в основания путем обработки их бикарбонатом натрия в водной среде с последующей экстракцией хлороформом. Из сгущенных хлороформных экстрактов осаждали алкалоиды в форме тартратов прибавлением 5 % раствора винной кислоты. Выпавшие тартраты смешивали с окисью магния. Выделенные таким образом основания экстрагировали хлороформом и после сгущения хлороформных экстрактов осаждали петролейным эфиром. Полученную смесь оснований растворяли в бензоле и хроматографировали на колонке с окисью алюминия. Бензолом элюировали алкалоиды эрготоксиновой группы, затем хлороформом – эрготамин.
Выделение алкалоидов группы эрготоксина. При упаривании бензольных элюатов выпадал кристаллический эрготоксин в виде комплексного соединения с бензолом с [a]D20 = –130°, –160° (с 1, хлороформ). Анализ его на бумажной хроматограмме в системе бензол – формамид показал, что он содержит в основном эргокристин, эргокриптина было меньше или он вовсе отсутствовал. Поскольку эргокорнин в этой системе располагался на хроматограмме на уровне эргокристина, дополнительно производился анализ косвенным путем: хроматографии на бумаге в системе бутанол – уксусная кислота – вода (4:1:5) подвергались аминокислоты, полученные в результате кислотного гидролиза эрготоксина. Присутствие валина в кислотном гидролизате должно было бы указать на содержание в исследуемом соединении эргокорнина. Результаты анализа показали, однако, что в большинстве полученных образцов комплекса эрготоксин – бензол эргокорнин отсутствовал; это хорошо согласуется с данными, полученными нами ранее, при исследовании алкалоидного состава дикорастущей спорыньи отечественного происхождения.
При перекристаллизации комплекса эрготоксин – бензол из ацетона выпадало крнсталлическое основание чистого эргокристина в соединении с ацетоном, [a]D20 = –165°, –187° (с 1, хлороформ). Это основание было обозначено в дальнейшем как эргокристин-ацетон. Выход его составлял около 60 % взятого для кристаллизации комплекса эрготоксин – бензол.
В ацетоновых маточниках оставался частично эргокристин, а также эргокриптин, если он первоначально обнаруживался в комплексе эрготоксин – бензол. Все наши попытки разделить эргокристин н эргокриптин фракционной кристаллизацией из разных растворителей не привело к положительным результатам.
Для разделения этих алкалоидов воспользовались методом фракционной кристаллизации их в виде солей ди-(n-толуил)-l-винной кислоты. Разделению подвергали первоначальный комплекс эрготоксин – бензол и ацетоновые маточники, полученные после отделения эргокристин-ацетона.
При фракционной кристаллизации в первую очередь выпадала кристаллическая нейтральная соль эргокристина, а затем эргокриптин в форме кислой соли.
Бромирование эргокриптина. Получают 2-бром-α-эргокриптин бромированием α-эргокриптина – основного алкалоида спорыньи эргокриптинового штамма [18]. Описано несколько методов бромирования с использованием различных бромирующих агентов: брома, N-бромсукцинимида (NБС), N-бромкапролактама, диоксандибромида, N-бромфталимида, гидротрибромида пирролидона-2, бромсахарина, гидротрибромида пиперидона-2 и др. Однако эти методы, как правило, либо сложны для промышленного исполнения, либо дают невысокий выход 2-бром-α-эргокриптина. Так, наиболее распространенный метод бромирования NБС дает выход продукта менее 50 %
Целью настоящей работы является поиск более эффективных методов бромирования В качестве исходного материала мы использовали индовидуальные α- и β-эргокриптины, а также более доступную смесь α- и β-эргокриптинов без предварительного их разделения, Эту задачу решали либо путем модернизации известного метода бромирования NБС, либо путем применения новых для этой реакции бромирующих агентов 2,4,4,6-тетрабромциклогексадиен-2,5-она (ТБЦГ), 2,2-дибром-5,5-диметилцикло-гександиона-1,3 (дибромдимедон), пербромида фенилтриметил-аммония (ФТМА) и «полимерного пербромида» на основе амберлита ИРА-402.
Экспериментальная часть
Контроль реакционных смесей осуществлялся хроматографически на пластинках «Silufol UV-254» в системе СН2Сl2 – диоксан – этиловый спирт – аммиак конц., 36:3:1:0,2, или на пластинках «DC-Alufolien» с нейтральной Al2O3 60F254 Typ E (ФРГ) в системе бензол – CHCl3, 1:1. Определение выхода 2-бромэргокриптинов при кинетических исследованиях проводили путем контроля на хроматографических пластинок на денситометре «Сromoscan 200» (Jouce Loebl).
Для изучения реакций бромирования в качестве исходного субстрата использовали техническую смесь изомеров α- и β-эргокриптинов состава 60:40 % соответственно, а также индивидуальные изомеры.
Полимерный пербромид на основе ионобменной смолы амберлит ИРА-402. Смешивают 10 г ионобменной смолы амберлит ИРА-402 с 5 % раствором KBr так, чтобы смола была полностью покрыта слоем раствора, и оставляют для набухания на ночь. Затем сливают верхний водный слой, смолу заливают свежим раствором KBr и приливают по каплям 2 мл брома при непрерывном перемешивании. Полученный бромирующий агент отфильтровывают, промывают водой, затем сухим диоксаном. Сушат сначала над CaCl2 в вакуум-эксикаторе, а затем над P2O5.
Бромирование эргокриптинов NБС. К нагретому до 60°С раствору 1 г (1,74 ммоля) смеси изомеров α- и β-эргокриптинов в 20 мл абс. диоксана (перегнанного над бензофенонкетилнатрием) в атмосфере азота и в темноте (или в зачерненной снаружи колбе) добавляют по каплям в течение 5 мин при перемешивании раствор 0,37 г (2,04 ммоля) NБС в 6,5 мл абс. диоксана. Реакционную смесь перемешивают в этих же условиях еще 70 мин, охлаждают, диоксан упаривают в вакууме при 40–50°С. Остаток растворяют в 30 мл СН2Сl2, полученный раствор промывают 20 мл 2 н. раствора Na2CO3. Водную фазу экстрагируют СН2Сl2 (2×10 мл). Объединенные органические фазы промывают 50 мл воды, сушат над Na2SO4, растворитель упаривают. Получают 1,1 г темного вязкого масла, которое очищают хроматографически на колонке (10×2 см) с Al2O3 III ст. активности, элюируя последовательно бензолом, смесью бензол – CHCl3, 4:1, 3:2, 1:1. Получают 625 мг (55 %) смеси α- и β-2-бромэргокриптинов. Полученный образец хроматографически идентичен с образцами, полученными при хроматографировании на силуфоле, Al2O3 («Merek»), а также методом ВЭЖХ.
Бромирование эргокриптинов 2,4,4,6-тетрабромциклогексадиен-2,5-оном. К нагретому до 60°С раствору 575 мг (1,0 ммоль) смеси α- и β-эргокриптинов в 20 мл абс. диоксана при перемешивании добавляют сразу раствор 410 мг (1,0 ммоль) тетрабромциклогексадиенона в 10 мл абс. диоксана. Реакционную смесь перемешивают в течение 30 мин при 60°С. Охлаждают, диоксан упаривают в вакууме, остаток растворяют в 30 мл СН2Сl2. Полученный раствор промывают 20 мл 5 % раствора NaHCO3, затем водой и сушат над Na2SO4. Растворитель упаривают в вакууме, остаток очищают хроматографически на колонке (10×2 см) с Al2O3 III ст. активности, элюируя последовательно смесью бензол – CHCl3, 3:1, 3:2, 1:1. Получают 382 мг (85 %) смеси α- и β-2-бромэргокриптинов.
Бромирование эргокриптинов пербромидом фенилтриметил-аммония. К раствору 575 мг (1,0 ммоля) смеси α- и β-эргокриптинов в 30 мл абс. СН2Сl2 (перегнанного над P2O5) при перемешивании добавляют 2 г мелко измельченной и предварительно высушенной при 120°С MgO. К полученной суспензии при перемешивании добавляют в течение 30 мин раствор 320 мг (0,85 ммоля) пербромида фенилтриметиламмония в 30 мл абс. СН2Сl2. В процессе добавления пербромида к реакционной смеси через каждые 5–7 мин вносят свежие порции MgO по 50–100 мг. Реакционную смесь фильтруют, осадок MgO тщательно промывают несколько раз 40 мл СН2Сl2, фильтрат промывают 20 мл 5 % раствора NaHCO3. Водный слой дополнительно экстрагируют СН2Сl2 (2×10 мл). Объединенные органические фракции промывают водой и сушат над Na2SO4. Растворитель упаривают в вакууме. Остаток растворяют в минимальном количестве бензола и наносят на хроматографическую колонку (16×2 см), заполненную Al2O3 IV ст. активности. Элюируют последовательно бензолом, смесью бензол – CHCl3, 5:1, 4:1, 3:1, 1:1. Получают 460 мг (70 %) смеси α- и β-2-бромэргокриптинов. Полученный образец идентичен образцам, полученным при хроматографировании на силуфоле, Al2O3 и методом ВЭЖХ.
Бромирование эргокриптинов «полимерным пербромидом» на основе амберлита ИРА-402. Растворяют 2 г α- и β-эргокриптинов в 40 мл абс. СН2Сl2, добавляют 4 г сухой MgO и при перемешивании в течение 0,5 ч вносят 6 г вышеуказанной смолы. Премешивают в течение еще 75 мин. После чего растовр отделяют от MgO и смолы. Раствор обрабатывают 40 мл 2 н. Na2CO3, затем водой (2×20 мл), органические фазы объдиняют, сушат Na2SO4, фильтруют и упаривают. Получают 2,1 г технического продукта, который очищают на колонке с Al2O3 III ст. активности в соотношении 1:30. Алкалоиды элюируют бензолом, смесью бензол – CHCl3 (5–30 %). Получают 0,4 г (20 %) смеси α- и β-2-бромэргокриптинов.
5.2 Синтез противорвотных лекарственных средств
Похожие работы

... рефлекторного характера. Однако практическое применение их ограничено, так как они действуют раздражающе на слизистую желудка и могут вызвать общетоксичекое действие. 3.2. ПРОТИВОРВОТНЫЕ СРЕДСТВА Если рвотные средства в настоящее время применяются редко, то борьба с рвотой, изнуряющей и обезвоживающей организм, необходимо довольно часто. Механизм рвотного акта обязательно должен учитываться ...
... порядке для применения с целью лечения, предупреждения или диагностики заболевания у человека или животного. Лекарственные средства можно классифицировать по следующим принципам: – терапевтическое применение (противоопухолевые, антиангинальные, противомикробные средства); ...
... , согласованного с национальными органами управления здравоохранением. В Российской федерации гомеопатические препараты подлежат такому же законодательному регулированию как и обычные лекарства. 2. Свойства лекарственных веществ Средства для наркоза. Для общего обезболивания в современной анестезиологии применяют различные лекарственные средства. В процессе подготовки к операции проводится ...
Бензоклидина гидрохлорид (оксилидин) Бенактизин (амизил) -пр. дифенилметана Темпидин - в составе темпалгина ------------------------------------------------------------------------------ СЕДАТИВНЫЕ ПРЕПАРАТЫ(I.) ПРЕПАРАТЫ БРОМА (в микстурах): Натрия бромид Калия бромид Бромкамфора Бромизовал (бромурал)(II.) РАСТИТЕЛЬНЫЕ ПРЕПАРАТЫ: Валерианы ...
0 комментариев