Создание и исследование новых лекарственных средств. Основное направление поиска
Роль компьютера при создании новых лекарственных средств
Определение спектра биологической активности с помощью программы PASS C&T (Prediction of Activity Spectra for Substances: Complex & Training)
Местноанестезирующие средства
Результаты эксперимента и их обсуждение
Спазмолитик
Проведение работы связано с определенными видами затрат
Навигация
Роль компьютера при создании новых лекарственных средств
Создание новых лекарственных веществ
73704
знака
10
таблиц
4
изображения
1.3 Роль компьютера при создании новых лекарственных средств
Ежегодно химики синтезируют, выделяют и характеризуют от 100 до 200 тысяч новых веществ. Многие из этих веществ проходят первичные испытания на выявление той или иной биологической активности. Этот этап поиска лекарственного вещества называют скринингом. Скрининг проводят в биологических лабораториях на живых клетках, микроорганизмах или кусочках живых тканей, на здоровых или специально задержанных животных: на мышах, крысах, морских свинках, собаках.
При этом из сотен веществ отбираются несколько наиболее активных препаратов, которые затем передаются на углубление испытания. Если высокая активность вещества подтверждается, то его всесторонне изучают для определения токсичности и побочных эффектов, при отсутствии или незначительности которых проводят кинетические испытания на людях.
Считается необходимым, чтобы все новые синтезируемые вещества были подвергнуты первичным испытаниям. Очевидно, что возможность испытать все новые соединения на все новые соединения на все нужные виды активности пока остается малореальной. В настоящее время существует возможность определения потенциала их биоактивности путем компьютерного анализа [1]. Достаточно лишь ввести в компьютер сведения о строении вещества. По окончании компьютерного анализа оператор получает рекомендации о целесообразности или нецелесообразности испытаний данного вещества на тот или иной вид активности. Скрининг экономит время, материалы и силы при аналоговом поиске лекарственных веществ.
В настоящее время также пользуются методом химического модифицирования структуры известных синтетических и природных лекарственных веществ. Этот метод является интуитивным, умозрительным. С его помощью исходя из аналогии двух структур биоактивность известного вещества как бы переносят на новое соединение.
Метод молекулярного моделирования в сочетании с рентгеноструктурным анализом позволяет установить стехереохимические особенности молекулы лекарственного вещества и биорецептора, конфигурацию их хиральных центров, измерить расстояние между отдельными атомами, группами атомов или между зарядами в случае цвиттер-ионных структур лекарства и биорецепторного участка его захвата. Получаемые таким образом данные позволяют более целенаправленно проводить синтезы биоактивных молекул с заданными на молекулярном уровне параметрами. Этот метод был успешно использован в синтезе высокоэффективных анальгетиков – аналогов морфина, а также для получения ряда лекарственных веществ, действующих на центральную нервную систему подобно природному нейромедиатору γ – аминомасляной кислоты. Широкое развитее получил метод комбинаторной химии.
Метод комбинаторной химии возник и стал быстро развиваться в 1990-х годах, как часть общей стратегии открытия новых лекарственных веществ.
Стратегия комбинаторной химии основана на недавней разработке нескольких революционных химических и биологических методов параллельного синтеза и испытания большого числа соединений. Была создана техника, позволяющая синтезировать в растворе или на твердых подложках от сотен до нескольких тысяч новых соединений в день и быстро их тестировать в виде смесей или после выделения индивидуальных веществ. В совокупности с автоматизацией синтез целых семейств вещества требует значительно меньше затрат реагентов при огромном росте производительности [9].
1.4 Молекулярное моделирование с помощью программы HyperChem
Молекулярное моделирование – сложная сеть различных наук, находящее применение в нанотехнологии, в молекулярной биологии, квантовой химии и биотехнологии.
Молекулярное моделирование молодая, востребованная и бурно развивающаяся наука.
На сегодняшний день методы квантовой химии и молекулярной динамики получили широкое распространение в численном моделировании электронной и атомной структур сложных систем молекулярных, кристаллических и переходных размеров. Это связано с технологическим развитием соответствующего математического обеспечения. Сейчас в мире функционирует достаточно много современных вычислительных комплексов, реализующих методы квантовой химии и молекулярной динамики. Использование многих из этих методов обеспечивается программой Hyper Chem для молекулярного моделирования.
HyperChem - комплексный программный продукт, предназначенный для задач молекулярного моделирования. Он включает в себя программы, реализующие методы молекулярной механики, квантовой химии и молекулярной динамики. Силовые поля, которые могут использоваться в HyperChem - это ММ+ (на базе ММ2), Amber, OPLS и BIO+ (на базе CHARMM). Реализованы полуэмпирические методы: расширенный метод Хюккеля, CNDO, INDO, MINDO/3, MNDO, AM1, PM3, ZINDO/1, ZINDO/S, а также возможности проведения неэмпирических расчетов и по теории возмущений Меллера-Плессета второго порядка.
HyperChem обладает развитыми средствами визуализации, которые могут использоваться как при подготовке входной информации (структуры молекулы), так и при анализе результатов, например, рассчитанных характеристик ИК- и УФ- спектров.
Расчётные методы оказывают неоценимую помощь в создании лекарственных средств. Молекулярное моделирование входит во все области знаний и находит себе применение, порой играя одну из главных ролей. Некоторые области химии немыслимы без молекулярного моделирования. В развитых странах моделирование является современным методом изучения микроструктур.
В настоящее время для изучения реакционной способности молекул используются приближения CNDO/2, MNDO, AM1, PM3.
Метод CNDO основан на приближении нулевого дифференциального перекрывания и поэтому является одним из простейших полуэмпирических методов. Из этого факта следуют ограничения применимости метода, который из-за обедненной расчетной схемы недостаточно корректно воспроизводит многие эффекты. С появлением более совершенных версий полуэмпирических методов МО приближение CNDO все реже применяется на практике. Так, в версии 7 программного продукта МОРАС данный метод не представлен. Тем не менее, во многих случаях для быстрой оценки электронных параметров полезно использовать схему CNDO, так как вследствие резкого уменьшения количества рассчитываемых интегралов с помощью этого метода можно исследовать более сложные объекты. В целом CNDO/2 дает надежные результаты при расчете электронных распределений и свойств, зависящих от них.
Основным калибровочным параметром в CNDO является резонансный интеграл. Он подбирается так, чтобы относительный порядок энергетических уровней занятых МО и коэффициенты разложения МО в ЛKAO наилучшим образом совпали с расчетами ab initio соединений обучающей выборки.
Общим достоинством всех перечисленных версий является прежде всего сравнительно малое время расчетов и меньшие размеры занимаемой оперативной памяти по сравнению с более точными приближениями. Это дает возможность как для быстрой оценки исследуемых объектов, так и для изучения более сложных молекул, требующих длительного времени расчета и больших объемов оперативной памяти. В целом приближение CNDO хорошо описывает электростатические эффекты и полярность связи. CNDO/2 может применяться для расчета дипольных моментов и зарядов по схеме Малликена и оценки равновесной геометрии.
Недостатки приближения CNDO являются следствием усечения расчетной схемы, которая не учитывает взаимодействия между перекрываниями орбитальных зарядов. В результате многие эффекты не воспроизводятся.
Метод MNDO был разработан на основе более строгого и сложного приближения NDDO. Это позволило существенно улучшить результаты расчетов при решении многих задач. Длительное время метод рассматривался в качестве основного полуэмпирического метода квантовой химии. Его возможности позволили с достаточной степенью надежности рассчитывать физико-химические свойства, электронные структуры и реакционную способность множества молекулярных систем.
Преимущество заключается в быстродействии (по сравнению с неэмпирическими методами) программ, в которых реализована схема MNDO. Это позволяет применять ее для исследования все более сложных объектов. Недостатки связаны с тем, что точность метода не может превышать точность тех экспериментальных данных, по которым проводилась параметризация.
В схеме MNDO используются 3 вида параметров.
Во-первых, варьируемые параметры, значения которых определяют с помощью оптимизационной процедуры.
Во-вторых, одноцентровые двухэлектронные интегралы, оценивающиеся из спектроскопических данных.
В-третьих, ряд зависимых параметров, необходимых для расчета двухцентровых двухэлектронных интегралов, оценивающиеся с помощью эмпирических схем.
В настоящее время область применения метода MNDO достаточно изучена. Зная особенности расчетной схемы MNDO, его преимущества и недостатки можно с успехом применять метод для решения многих задач.
Хотя в целом метод MNDO имеет существенные преимущества перед СNDO, в некоторых случаях метод дает серьезные сбои. Это в первую очередь касается расчетов молекул с водородными связями, барьеров внутреннего вращения в p-сопряженных системах и расчетов четырехчленных циклов. Поэтому в рамках метода MNDO были разработаны модифицированные варианты.
Для расчета характеристик систем с водородными связями были разработаны методы MNDO/Н и MNDO/М, которые лучше воспроизводят экспериментальные значения энергии водородных связей в комплексах.
Удовлетворительное описание водородных связей позволило широко использовать модифицированные варианты для исследования биологических объектов.
Таким образом, полуэмпирические квантовохимические методы можно использовать с большим практическим выходом для изучения реакционной способности различных химических соединений.
| Достоинства метода MNDO | Недостатки метода MNDO |
| 1.Быстродействующий метод, позволяющий изучать строение и свойства сложных молекул. 2.Учитывается ориентация р-орбиталей и правильно описывается отталкивание неподеленных электронных пар. 3.Значительное расширение круга доступных для расчета соединений. 4.Более корректное описание последовательности верхних молекулярных уровней. 5.Может использоваться для интерпретации фотоэлектронных спектров. 6.Преимущества проявляются в большей степени в расчетах более полярных молекул. 7.Удовлетворительно описывается строение радикалов, дает разумные результаты для катионов органических соединений. 8.Более точно рассчитываются валентные углы. 9.Более надежный расчет частот колебаний, протонных эффектов и электронного сродства. 10.При изучении химических реакций правильно описывается ППЭ и ПС. | 1.Точность метода не может превышать точности экспериментальных данных. 2.Электронная корреляция учитывается дважды. Правильнее было бы внесение корреляционных поправок. 3.Недооцениваются эффекты взаимодействия через пространство. 4.Ограничения возможности расчета соединений третьего и более высоких периодов (в частности с SO и SO2-группами) вследствии пренебрежения d-орбиталями. 5.Некорректное описание водородных связей. 6.Неверен расчет барьеров внутреннего вращения в сопряженных молекулах. 7.Недооценивается энергия трехцентровых связей. 8.Неудовлетворительная оценка спиновых плотностей и констант сверхтонкого расщепления электронного парамагнитного резонанса. 9.Завышается стабильность радикалов. 10.Потенциалы ионизации атомов III периода с сильно выраженным S-характером завышены вследствие применения приближения замороженного остова. 11.Плохо воспроизводится спиновая плотность в радикалах. 12.Энтальпия одноатомных ионов может значительно отличаться от экспериментальных данных. 13.Ограниченная воспроизводимость свойств неорганических молекул. Ошибка расчета составляет для энтальпии - 10 ккал/моль, потенциала ионизации - 1 эВ, длин связей - 0,07A |
Похожие работы
... на фармакологический эффект, усложняет процесс изыскания новых Л В. Тем не менее современные методы исследования позволили определить предпосылки решения этой важной проблемы. 7 Предпосылки создания новых лекарственных веществ Изыскание новых ЛВ осуществляют различными путями. Ведущим направлением являются исследования в области модификации структуры известных природных БАВ. Одним из ...
... процесс разделения нестабильных веществ можно проводить в холодильной камере. Выделенное соединение подвергают структурному химическому исследованию, а затем изучают его фармакологическое действие. Получение лекарственных веществ методом культуры тканей высших растений В нашей стране заготавливаются десятки тысяч тонн ЛРС. Однако потребность в БАВ, содержащихся в растениях, с каждым годом ...
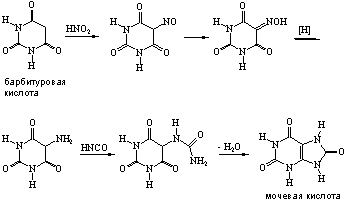
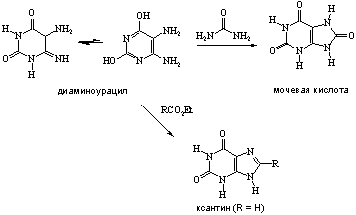
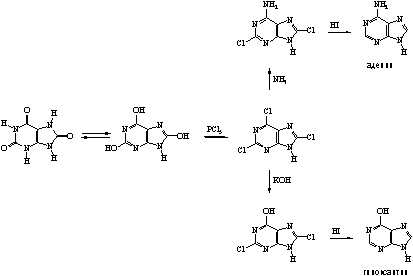
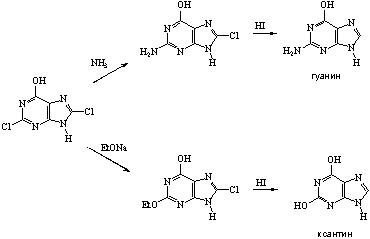
... 6. При взаимодействии продуктов замещения с иодистоводородной кислотой происходит гидродехлорирование атомов хлора, сохранившихся в молекуле после реакции с нуклеофилом, что приводит к образованию двух важных производных пурина - 6-оксипурина (гипоксантина) и 6-аминопурина (аденина). После замещения атома хлора в положении 6 можно провести замещение атома хлора в положении 2. Различие в ...

... с высокой пропускной способностью (HTS-метод). Сегодня HTS-метод (High Throughput Screening) повсеместно используется в фармацевтической индустрии для открытия новых лекарственных средств. С помощью высокоскоростной компьютеризованной технологии сотни тысяч веществ проверяются на активность относительно исследуемой молекулы, предназначенной для взаимодействия[8]. 2. Применение компьютерного ...
0 комментариев