Продукт гидролиза при дегидратации и последующем озонолизе даёт смесь уксусного альдегида и ацетона
Осуществите превращения, все продукты назовите, укажите условия химических превращений
Углеводы (моносахариды, полисахариды). Гликозиды. Крахмал (амилоза, амилопектин), декстран (полиглюкин)
Пектиновые вещества. Эфиры целлюлозы метил-, карбоксиметил- и натрийкарбоксиэтилцеллюлоза). Растительные камеди
Нуклеозиды, нуклеотиды, нуклеиновые кислоты
Липиды
Алкалоиды
Воски. Спермацет
Строение, физические и химические свойства высокомолекулярных полимеризационных и поликонденсационных полимеров, используемых в фармации
Современные физико-химические методы в анализе и идентификации органических соединений
Масс-спектрометрический метод
Общие правила и порядок работы в химической лаборатории. Правила техники безопасности
Напишите строение участка РНК-АУ (Аденин-Урацил). Укажите сложноэфирные и гликозидные связи
На примере пиразола покажите строение пиррольного и пиридинового атомов азота. С помощью химических реакций докажите амфотерный характер пиразола
Напишите схему получения 2,6-дигидроксипурина (ксантина) из мочевой кислоты. Покажите таутомерные превращения ксантина
Навигация
Масс-спектрометрический метод
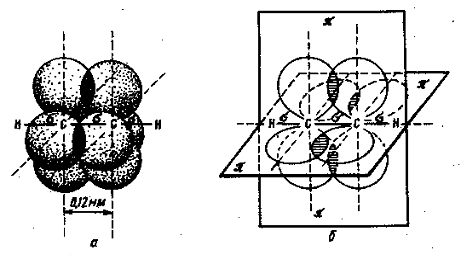
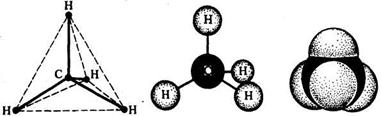
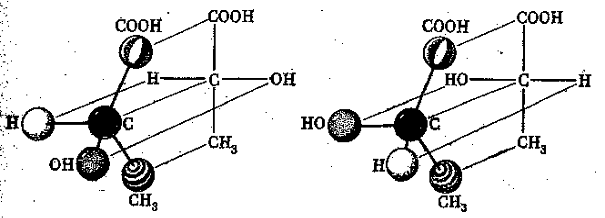
Органическая химия
155076
знаков
2
таблицы
73
изображения
8.2 Масс-спектрометрический метод
Применение масс-спектрометрии. Масс-спектрометрию широко применяют в разл. областях науки и техники: в химии и нефтехимии, физике, геологии, биологии, медицине, в пром-сти полимеров, в лакокрасочной и хим. пром-сти, в произ-ве полупроводников и сверхчистых материалов, в ядерной технике, в с. х-ве и ветеринарии, в пищ. пром-сти, при анализе продуктов загрязнения окружающей среды и мн. др. Большие успехи достигнуты при анализе биологически важных в-в; показана возможность структурного анализа полисахаридов с мол. м. до 15000, белков с мол. м. до 45000 и т.д. Масс-спектрометрия нашла применение как экспрессный метод газового анализа в медицине; принципы масс-спектрометрии лежат в основе устройства наиб. чувствит. течеискателей. Отечеств. масс-спектрометры, выпускаемые для разл. целей, имеют индексы: для исследования изотопного состава - МИ, для исследования хим. состава - MX, для структурного анализа - МС. Macс-спектрометрия в органической химии позволяет измерить точную мол. массу и рассчитать элементный состав исследуемого в-ва, установить хим. и пространств. строение, определить изотопный состав, провести качеств. и количеств. анализ сложных смесей орг. соединений. Одна из важнейших задач - нахождение зависимости между характером масс-спектра и строением исследуемой орг. молекулы. При ионизации орг. молекулы образуется мол. ион, в к-ром далее происходят процессы гетеро- и гомолитич. разрыва связей или разрыва связей с перегруппировкой молекулы и образование осколочных ионов, к-рые в свою очередь могут подвергаться дальнейшему распаду. Последоват. распады ионов, устанавливаемые из масс-спектра, наз. направлениями или путями распада. Направления распада - важная характеристика каждого класса соединений. Совокупность всех направлений распада составляет характерную для каждого орг. соед. схему фрагментации. Если масс-спектр прост, схема фрагментации сводится к одному пути распада, напр. при распаде мол. иона СН3ОН+ последовательно образуются ионы СН2=ОН+ и Н—С=О+. В случае сложных масс-спектров схема фрагментации отвечает многим, часто перекрывающимся направлениям распада, напр. схема фрагментации полипептида:
Мол. ион пептида распадается в результате разрыва связей СН—СО, СО—NH, NH—СН и СН—R с образованием осколочных ионов соотв. Аn и Хn, Вn и Yn, Сn и Zn, Sn и Rn (n - номер аминокислотного остатка в пептидной цепи), к-рые далее распадаются таким же образом. Общее кол-во пиков ионов в таком спектре может достигать неск. сотен. Кол-во фрагментов определяется строением исследуемой молекулы, запасом внутр. энергии мол. и осколочных ионов и промежутком времени между образованием иона и его детектированием. Поэтому при интерпретации масс-спектров необходимо учитывать как условия измерений (энергию ионизирующих электронов, ускоряющее напряжение, давление паров в ионном источнике, т-ру ионизац. камеры), так и конструктивные особенности прибора. При макс. стандартизации условий измерений удается получать достаточно воспроизводимые масс-спектры. Сравнение масс-спектра исследуемой системы со спектром, имеющимся в каталоге, -наиб. быстрый и простой способ структурного анализа, идентификации в-в при определении загрязнения окружающей среды, контроле продуктов питания человека и животных, изучении процессов метаболизма лек. препаратов, в криминалистике и т.д. Однако идентификация лишь на основании масс-спектра не может быть однозначной, напр. не все изомерные в-ва образуют различающиеся масс-спектры. В условиях масс-спектрометрии часть возбужденных ионов распадается после выхода из ионного источника. Такие ионы наз. метастабильными. В масс-спектрах они характеризуются уширенными пиками при нецелочисленных значениях т/z. Один из методов изучения таких ионов - спектроскопия масс и кинетич. энергий ионов. Изучение распада метастабильных ионов проводят на приборах, у к-рых магн. анализатор предшествует электрическому. Магн. анализатор настраивают таким образом, чтобы он пропустил метастабильный ион, к-рый при определенном напряжении на электрич. анализаторе проходит в детектор. Если такой ион распадается в пространстве между анализаторами, то образующиеся вторичные ионы не могут пройти через электрич. анализатор при установленном напряжении из-за недостатка энергии. Для попадания вторичных ионов в детектор изменяют напряжение электрич. анализатора. Это напряжение связано с массой вторичного иона соотношением m2 = Е2m*/Е0, где m* - метастабильный ион, m2 - вторичный ион, Е0 и Е2 - начальное и конечное напряжение электрич. анализатора. Таким образом определяются массы всех ионов, образующихся при распаде метастабильных ионов и устанавливаются тем самым схемы их фрагментации. Если в области между двумя анализаторами создать область повыш. давления (установить камеру столкновений, заполненную инертным газом), то в результате соударений ионов с молекулами газа их внутр. энергия будет увеличиваться и, следовательно, увеличится вероятность образования вторичных ионов. Такой метод, наз. тандемным, используют для структурного анализа индивидуальных компонентов сложных смесей без предварит. разделения. Наряду со структурными исследованиями масс-спектрометрию применяют для количеств. анализа орг. в-в. Количеств. анализ основан на определении интенсивностей пиков ионов с определенным значением т/z. Его проводят хромато-масс-спектрометрически (см. Хромато-масс-спектрометрия) или в системе прямого ввода. Для повышения точности определения применяют внутр. стандарты, в качестве к-рых используют меченые соед. или соед. близкие по строению к исследуемым, напр. гомологи. В последнем случае необходимо построение калибровочных кривых. Измерение содержания исследуемого в-ва проводят с учетом кол-ва добавляемого стандарта по отношению площадей пиков, соответствующих определяемому в-ву и внутр. стандарту. Погрешность метода b7%, предел определения 0,01 мкг/мл. Лучшие результаты дает применение меченых соед.; при этом отпадает необходимость в построении калибровочных кривых. Количеств. определение труднолетучих в-в проводят в системе прямого ввода, детектируя их по одному или неск. ионам, характерным для исследуемого соединения. По мере плавного повышения т-ры испарителя происходит испарение и частичное фракционирование исследуемых в-в. Т. обр., для каждого в-ва получают кривую испарения, площадь под к-рой прямо пропорциональна кол-ву соед., внесенного в масс-спектрометр. Абс. чувствительность метода, наз. методом интегрирования ионного тока, 10-7 г. Достоинство метода - отсутствие необходимости предварит. очистки исследуемых в-в. При исследовании соед. с электроф. группировками, изомерных орг. молекул, полимеров, азокрасителей, биологически активных в-в применяют масс-спектрометрию отрицательно заряженных ионов. Эти ионы обладают меньшим запасом внутр. энергии, чем положительно заряженные ионы, поэтому в масс-спектрах дают интенсивные пики мол. ионов и малое кол-во осколочных ионов.
9 Основные приёмы техники лабораторной работы (экстракция, выделение, очистка, определение температуры плавления, различные виды перегонки, кристаллизация)
ВЫДЕЛЕНИЕ. Синтезируемое вещество, получаемое в результате какой-либо реакции, обычно находится в реакционной смеси совместно с другими веществами (другие продукты, получающиеся по основному уравнению реакции; побочные продукты реакции; растворитель, в котором проводилась реакция). Поэтому всегда возникает задача выделения нужного вещества из весьма сложной подчас смеси. Иногда такое выделение удается не сразу; часто вначале вещество выделяют не вполне чистым и только в результате дальнейшей обработки получают чистый продукт.
Между методами выделения вещества из сложной реакционной смеси и методами его последующей очистки нет резкой разницы. Обычно и в том и в другом случаях используют различие в растворимости и в летучести веществ, имеющихся в смеси. Кроме того, для очистки пользуются различной способностью разных веществ поглощаться адсорбентами, например активным углем.
Использование различия в растворимости органических веществ лежит в основе выделения и очистки их методами кристаллизации и экстракции, а различия в летучести - в основе очистки перегонкой.
Кристаллизация
При очистке органического вещества кристаллизацией задача заключается в том, чтобы создать благоприятные условия для выделения данного вещества в кристаллическом состоянии из пересыщенного раствора и в то же время удержать в растворе сопутствующие примеси.
Из двух методов получения пересыщенных растворов - путем испарения части растворителя и путем охлаждения растворов, насыщенных при нагревании, - предпочитают пользоваться последним. При кристаллизации через охлаждение пользуются такими растворителями, в которых растворимость кристаллизуемого вещества резко изменяется с температурой. Существенной является также способность растворителя хорошо растворять примеси; чем больше разница в величинах растворимости основного продукта и примесей, тем легче осуществляется очистка. Нужно отметить, что загрязнения могут сильно влиять на скорость кристаллизации и на полноту выделения кристаллизуемого вещества из раствора. Иногда в присутствии значительного количества примесей кристаллизация может вообще не наступить, а если и удается добиться выделения кристаллов, то потери вещества в маточном растворе оказываются слишком большими. Поэтому во многих случаях к очистке вещества путем кристаллизации следует прибегать лишь после освобождения его от значительной части примесей другими способами, например перегонкой.
В качестве растворителя при кристаллизации наиболее часто применяют воду, этиловый спирт, метиловый спирт, бензин, бензол, петролейный эфир, этиловый эфир, уксусноэтиловый эфир, ледяную уксусную кислоту, хлороформ. Для труднорастворимых соединений используют также нитробензол, пиридин, фенол, анилин.
Большое значение для успеха работы имеет правильный выбор растворителя. При выборе растворителя необходимо учитывать состав и строение растворяемого вещества. Так, вещества, содержащие гидроксильные группы, в большинстве случаев более или менее хорошо растворяются в воде. Увеличение длины углеводородной цепи, например в высших спиртах, резко уменьшает растворимость в воде, но увеличивает растворимость в спиртах и углеводородах.
Окончательный выбор растворителя можно произвести лишь опытным путем. Для этого берут несколько пробирок, помещают в них небольшое количество вещества (например, по 0,2 г), прибавляют 0,5-1 мл различных растворителей и нагревают до полного растворения. Наиболее подходящим будет тот растворитель, из которого по охлаждении выделяется хорошо образованные кристаллы в небольшом количестве. Если в одном из растворителей вещество растворяется очень хорошо, а в другом - плохо, то следует испытать их смесь. Часто применяют смесь спирта с водой, ацетона с водой, эфира с бензолом.
Растворимость вещества в выбранном растворителе на холоду не должна быть слишком большой, так как это приводит к чрезмерно большим потерям вещества в маточном растворе. Кроме того, в этом случае пришлось бы работать с небольшими объемами жидкости, что привело бы к увеличению механических потерь (размазывание по стенкам, неполнота стекания и т. п.). В случае малой растворимости работа осложняется необходимостью оперировать со слишком большими объемами растворов.
Самая кристаллизация проводится следующим образом. Подлежащие очистке вещество помещают в колбу, обливают небольшим количеством растворителя, нагревают до кипения и затем добавляют понемногу новые порции растворителя (доводя после этого раствор снова до кипения) до полного растворения вещества*1. Чтобы растворитель не испарялся, колбу соединяют с обратным холодильником и растворитель приливают через трубку холодильника. Нагревание обычно ведут на водяной бане, за исключением тех случаев, когда работают с высококипящими растворителями; при приливании горючих растворителей горелку отставляют.
Полученный концентрированный раствор необходимо профильтровать (для удаления нерастворимых примесей, волокон фильтровальной бумаги и других загрязнений). Фильтрование ведут с отсасыванием через достаточно большую воронку Бюхнера (Рис. 7), вставленную в толстостенную коническую колбу для отсасывания*2. Если вещество при охлаждении кристаллизуется очень легко, то в случае концентрированных растворов кристаллизация начинается в самой воронке, ее отверстия забиваются и фильтрование прекращается. Чтобы избежать этого, растворитель берут в избытке (небольшом), а воронку перед фильтрованием осторожно подогревают пламенем горелки.
Во избежание кристаллизации во время фильтрования можно также пользоваться воронкой для горячего фильтрования. Эта воронка имеет двойные стенки, между которыми наливается вода, подогреваемая горелкой. Внутрь этой воронки вставляется обычная стеклянная воронка с фильтром.
При работе с легколетучими растворителями фильтрование с отсасыванием приводит к слишком большим потерям растворителя за счет испарения. В этих случаях следует фильтровать через обычную коническую воронку со вставленным в нее складчатым фильтром из неплотной фильтровальной бумаги; для уменьшения испарения растворителя воронку накрывают часовым стеклом (выпуклой стороной книзу).
Для получения хорошо образованных кристаллов необходимо охлаждать раствор медленно, при полном покое. Часто при попадании горячего фильтруемого раствора в холодный приемник наблюдается быстрое выделение обычно плохо образованных кристаллов. В этом случае профильтрованный раствор необходимо снова нагреть до растворения кристаллов и оставить медленно охлаждаться. Во многих случаях кристаллизация наступает очень медленно. Для ускорения ее прибегают к трению стеклянной палочкой о стенки сосуда или к внесению "затравки" (кристаллик ранее полученного препарата того же вещества). Как только кристаллизация начнется, раствор оставляют стоять в покое.
Для более полного выделения кристаллов из маточного раствора часто прибегают к его охлаждению при помощи охлаждающих смесей или же ставят сосуд с раствором в холодильный шкаф. Растворимость большинства веществ при низких температурах уменьшается, и поэтому путем охлаждения достигается большая полнота выделения кристаллизуемого вещества из раствора. Однако нужно учитывать, что понижение температуры может уменьшить скорость роста кристаллов, что особенно заметно в случае вязких жидкостей.
Для удобства извлечения образовавшихся кристаллов рекомендуется проводить кристаллизацию в конических колбах или в стаканах, но не в обычных плоскодонных колбах. При работе с летучими растворителями пользуются только коническими колбами, которые во избежание испарения растворителя накрывают часовым стеклом (выпуклой стороной кверху). Ни в коем случае не следует колбу с горячим раствором плотно закрывать пробкой: при охлаждении в колбе создается вакуум (вследствие конденсации паров) и она может быть раздавлена атмосферным давлением.
Для удаления из раствора окрашенных и смолообразных примесей, затрудняющих кристаллизацию и загрязняющих получаемые кристаллы, с успехом применяют активный уголь (крупнопористые сорта). Уголь, во избежание внезапного вскипания жидкости, следует вносить в несколько охлажденный раствор, когда все подлежащее кристаллизации вещество растворилось. После прибавления активного угля раствор нагревают до кипения, кипятят несколько минут и затем фильтруют.
Уголь прибавляют в количестве, необходимом для полного обесцвечивания раствора, избегая в то же время большого избытка. Для этого уголь вносят небольшими порциями, после внесения каждой из них раствор кипятят и затем дают ему несколько отстояться, чтобы можно было установить, в достаточной ли мере удалены смолистые и окрашенные примеси. Так поступают до тех пор, пока не будет достигнут нужный эффект очистки.
Иногда частицы слишком мелко растертого угля проходят сквозь фильтр и загрязняют фильтрат. Этот недостаток может быть устранен предварительным взмучиванием угля в воде и декантацией (после отстаивания) взвешенный мелких частиц. При работе с неводными растворителями промытый уголь высушивают на водяной бане.
Если раствор фильтруется плохо и фильтр забивается, то иногда полезно прибавить к углю немного мелких древесных опилок. В тех случаях, когда после осветления углем вещество предполагают подвергнуть анализу (элементарному), нужно особенно тщательно следить, чтобы частицы угля не попали в фильтрат. Лучше всего перед анализом перекристаллизовать вещество еще раз, уже без применения активного угля.
Полученные кристаллы отделяют от маточного раствора фильтрованием с отсасыванием на воронке Бюхнера или, в случае жидкостей, действующих на бумагу, - на воронках с фильтровальными пластинками из пористого стекла. Размеры воронки должны соответствовать количеству отсасываемого вещества; применение воронок слишком больших размеров приводит к ненужным потерям вещества. Для отфильтровывания очень малых количеств кристаллов (порядка 0,1 г и менее) пользуются обычной маленькой стеклянной воронкой, в которую вставляют стеклянную палочку с расплюснутым концом - "пуговкой". Для приготовления такой "пуговки" конец тонкой стеклянной палочки нагревают до размягчения и затем прижимают ко дну ступки, к керамиковой плитке и т. п. Стеклянная палочка должна быть настолько тонкой и длинной, чтобы она свободно входила в трубку воронки и конец ее выдавался немного снизу. На "пуговку" кладут кружок фильтровальной бумаги немного большего диаметра, так чтобы он плотно прилегал к стенкам воронки (рис. 8). Воронку вставляют или в маленькую колбу для отсасывания, или в укрепленную в штативе пробирку для отсасывания.
Для того чтобы фильтр плотно прилегал к стенкам воронки, его полезно смочить водой, отсосать воду, промыть небольшим количеством спирта и под конец - тем растворителем, который нужно будет отсасывать.
Фильтр, вкладываемый в воронку Бюхнера, должен быть несколько меньшего диаметра, чем воронка, и, полностью закрывая все отверстия дна воронки, не должен в то же время прилегать к ее стенкам. Перед фильтрованием фильтр нужно смочить растворителем и затем включить насос. Кристаллы из сосуда, в котором производилась кристаллизация, переносят на фильтр с помощью стеклянной палочки. Кристаллы, приставшие к стенкам сосуда, смывают небольшими порциями отфильтрованного маточного раствора. Для более полного удаления маточного раствора часто бывает полезным отжать кристаллы на фильтре (не прекращая отсасывания) при помощи шпателя, пестика или стеклянной пробки.
После того как маточный раствор отфильтрован, не следует просасывать воздух через кристаллы, так как растворитель при этом испаряется и содержащиеся в нем примеси остаются на кристаллах. Для удаления маточного раствора, захваченного кристаллами, их необходимо промыть возможно малым количеством холодного растворителя. Для этого перекрывают отсасывание, смачивают осадок растворителем, дают немного постоять, чтобы осадок равномерно пропитался жидкостью, и отсасывают. Эту операцию повторяют еще раз или два (но не более). Большинство органических веществ довольно хорошо растворяется даже в холодных растворителях; поэтому хорошее промывание осадка при минимальных потерях вещества, требует от работающего известного навыка.
В маточных растворах и промывных жидкостях часто остается такое количество вещества, которым не следует пренебрегать. В таких случаях надо отогнать часть растворителя и снова довести раствор до кристаллизации. Полученные при этом кристаллы обычно бывают менее чистыми, чем первая порция, и их следует перекристаллизовать еще раз.
Высушивание осадка. По окончании промывания осадок вместе с фильтром вынимают из воронки, кладут на сложенную в несколько раз фильтровальную бумагу, удаляют пинцетом фильтр и отжимают осадок между листьями фильтровальной бумаги. В большинстве случаев для окончательного удаления растворителя оказывается достаточным простое высушивание осадка на воздухе при комнатной температуре. С этой целью отжатый осадок рассыпают тонким слоем на листе фильтровальной бумаги, покрывают (для защиты от пыли) другим листом фильтровальной бумаги и оставляют до полного высыхания.
Иногда высушивание препарата можно ускорить, нагревая его в сушильном шкафу. Этот способ следует, однако, применять с осторожностью и только в случае вещества с высокой температурой плавления, так как небольшая примесь еще не удаленного растворителя может существенно снизить температуру плавления и вещество может при нагревании расплавиться.
Вещества гигроскопические нужно сушить в эксикаторе. В качестве водуотнимающих средств в эксикатор помещают окись алюминия, хлористый кальций, концентрированную серную кислоту или фосфорный ангидрид. Следует особенно рекомендовать применение окиси алюминия и хлористого кальция.
Окись алюминия очень энергично поглощает воду и может связать до 15-20% воды от собственного веса. Она легко регенерируется путем нагревания до 175° в течение 6 час. с последующим охлаждением в эксикаторе. Хлористый кальций несколько уступает окиси алюминия (а также и серной кислоте) по способности связывать воду, но он является легко доступным, дешевым продуктом, легко регенерируется путем прокаливания и свободен от тех недостатков, которые, как указано ниже, присущи серной кислоте.
Серная кислота, хорошо поглощая воду, одновременно поглощает и пары органических веществ; в результате их постепенного окисления она восстанавливается до сернистого ангидрида, который может реагировать с высушиваемом веществом. Другим недостатком применения серной кислоты является возможность ее расплескивания при переноске эксикатора, в результате чего брызги кислоты могут падать на дно сосуда с высушиваемым веществом. Чтобы кислота не расплескивалась, на дно эксикатора насыпают достаточно толстым слоем битое стекло. Для того чтобы установить момент, когда серная кислота станет непригодной в качестве высушивающего средства, в ней растворяют (перед помещением в эксикатор) сернокислый барий (из расчета 18 г сернокислого бария на 1 л концентрированной серной кислоты). Выпадение осадка сернокислого бария указывает на то, что кислота уже непригодна для сушки и должна быть заменена свежей. Нужно отметить, что при вакууме порядка 1 мм серная кислота несколько летуча и поэтому ее не рекомендуется применять в вакуум-эксикаторах при больших разрежениях.
Фосфорный ангидрид связывает воду очень энергично, но при этом на его поверхности образуется сиропообразная корочка, препятствующая дальнейшему поглощению воды, что является существенным недостатком.
Определение температуры плавления
Температурой плавления считается температура, при которой замечается первое появление жидкой фазы. Разность между температурой, при которой появляется жидкая фаза, и температурой полного расплавления вещества, не должна превышать 0,5 °С. Незначительные загрязнения вещества иногда сильно понижают температуру его плавления, и оно происходит в более широком интервале температур. Такое явление используют для установления идентичности двух веществ с одинаковой температурой плавления. Для этого смешивают равные количества двух веществ. Если температура плавления этой "смешанной" пробы остается неизменной, то делают заключение об идентичности обоих веществ. Понижение же температуры плавления пробы служит признаком неидентичности. Оценка идентичности исследуемого вещества по температуре плавления ''смешанной" пробы является настолько общепринятой, что этот прием считается достаточным для вынесения окончательного решения.
Многие органические вещества плавятся с разложением, которое обычно обнаруживается по окрашиванию расплава или выделению газа. В качестве характеристики веществ, которые плавятся с разложением, в справочнике приведена величина температуры плавления с дополнением "разл.". Существуют различные приборы для определения температуры плавления органических веществ.
Наиболее простой прибор для определения температуры плавления состоит из круглодонной колбы, заполненной соответствующей обогревающей жидкостью и имеющей боковые отверстия для испарения этой жидкости. В колбу вставлена пробирка с термометром, к которому прикреплен капилляр с веществом.
В качестве теплопередающей среды используют воду, серную кислоту, силиконовое масло и др. В данном приборе температуру плавления органического кристаллического вещества определяют в капилляре, запаянном с одного конца. Испытуемое вещество растирают в ступке. Открытым концом капилляра набирают в него немного вещества и бросают его запаянным концом вниз в стеклянную трубку длиной 60-80 см, поставленную вертикально на лабораторный стол. Эту операцию наполнения повторяют несколько раз до получения в капилляре хорошо уплотненного столбика вещества высотой 3-4 мм. Наполненный капилляр закрепляют резиновым кольцом на термометре так, чтобы проба вещества находилась на уровне ртутного шарика термометра. Нагревают прибор электрической плиткой. Когда исследуемое вещество начинает заметно плавиться либо сжиматься и мокнуть, плитку убирают. Началом плавления считают появление первой капли в капилляре, а окончанием – исчезновение последних кристалликов вещества.
В рабочем журнале отмечают температуру плавления вещества и все изменения, происходящие с ним в процессе нагревания: перемену окраски, разложение и т.п.
Разделение органических жидкостей простой перегонкой
Перегонкой называется процесс, в ходе которого вещество нагревают в соответствующей аппаратуре до кипения и образовавшийся пар конденсируют. Целью перегонки является разделение на компоненты и очистка жидких летучих веществ, имеющих различные температуры кипения (при этом во время процесса наблюдают за температурой). Простую перегонку целесообразно применять для жидкостей с температурой кипения от 40 до 150 °С, загрязненных небольшими количествами примесей, имеющих ничтожное давление пара при температуре кипения очищаемой жидкости, или когда разница в температурах кипения веществ, входящих в состав разделяемой смеси значительна (не менее 80-100 °С).
Если перегоняемая жидкость кипит не выше 120- 130 °С, то применяют проточное водяное охлаждение. При перегонке жидкостей, кипящих выше этой температуры, применяют воздушный холодильник.
При перегонке индивидуальных веществ их температура кипения остается постоянной в течение всей перегонки. Если перегоняется смесь двух веществ, температуры которых различаются значительно, то вначале отгоняется жидкость, имеющая более низкую температуру кипения. Если же температура кипения начинает возрастать, то это означает, что начинает отгоняться другая жидкость, имеющая более высокую температуру кипения, чем первая. В процессе перегонки второй жидкости также устанавливается постоянная температура. Таким образом, меняя приемники, можно собрать несколько фракций, в первых будет преобладать низкокипящая часть перегоняемой смеси, а в последних - высококипящая.
Если перегоняемая смесь состоит из компонентов, температуры кипения которых близки и которые не образуют азеотропных смесей, то применяют дробную или фракционную перегонку. Для этого обычно используют дефлегматоры или ректификационные колонки.
Для веществ, разлагающихся до- или при температуре кипения при атмосферном давлении или имеющих высокую температуру кипения, применяют перегонку при пониженном давлении.
Схема установки для простой перегонки показана на рис.1. Установка состоит из круглодонной колбы 1, дефлегматора 2, термометра 3, нисходящего холодильника Либиха 4, алонжа 5 и приемника 6. Ртутный шарик термометра должен находиться примерно на 0,5 см ниже отводной трубки насадки.
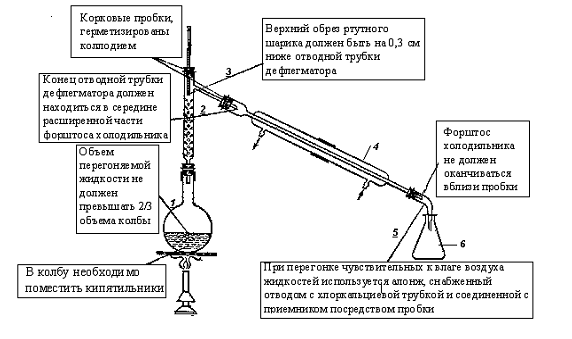
Перед заполнением прибора измеряют объем или вес жидкости, предназначенной для простой перегонки. Заливают жидкость в колбу 1 не более чем на 2/3 её объема. Для равномерного кипения в колбу помещают "кипелки". Включают воду для охлаждения. В качестве теплоносителя используют “баню”, соответствующую температуре кипения наиболее высококипящего компонента перегоняемой смеси. В качестве нагревательного прибора используют электроплитки только с закрытой спиралью. Баню нагревают до температуры, при которой вещество перегоняется с нормальной скоростью (из холодильника поступает в приемник 30-40 капель конденсата в минуту). Контроль за температурой в бане осуществляют с помощью термометра, помещённого в неё. Разность температуры бани и температуры кипения определяется рядом факторов: летучестью отгоняемого вещества, его количеством, конструкцией установки (наличие дефлегматора, высота горла колбы и насадки).
Температура бани, как правило, превышает температуру кипения перегоняемого вещества на 20- 30 °С.
Очистка органических жидкостей перегонкой с водяным паром
Сущность очистки органических жидкостей перегонкой с водяным паром заключается в том, что высококипящие жидкости, не смешивающиеся или мало смешивающиеся с водой, улетучиваются вместе с водяным паром при пропускании его через эти жидкости, затем они вместе с паром конденсируются в холодильнике.
Прибор, используемый при перегонке с водяным паром, изображен на рис. 2. Пар образуется в паровике 1 (вместо него пригодна и колба). Предохранительная трубка 2 служит для выравнивания давления. Паровик заполняют водой приблизительно на половину.
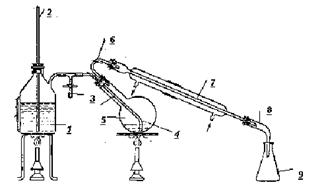
Рис. 2. Прибор для перегонки с паром
Пар через проводящую трубку 4 входит в перегонную колбу 5, в которой находится разделяемая смесь. Обычно эту колбу нагревают. Дистиллят через трубку 6 или насадку Вюрца с термометром поступает в холодильник 7, конденсируется и через алонж 8 стекает в приемник 9.
На тройник 3 надевают короткую резиновую трубку с зажимом. Зажим остается открытым до начала перегонки. В колбу помещают вещество, собирают прибор и подогревают парообразователь, предварительно поместив в него "кипелки". Одновременно подогревают колбу. Как только начнет образовываться пар, резиновую трубку, надетую на тройник, закрывают зажимом.
Спустя некоторое время в приемнике собирается эмульсия, расслаивающаяся при стоянии. Перегонку заканчивают, когда в холодильнике будут образовываться капли чистого дистиллята (воды). Затем открывают зажим на тройнике (если таковой отсутствует, то просто вынимают пароподводящую трубку 4 из перегонной колбы) и выключают парообразователь. Получающиеся в приемнике два слоя: воду и органическое вещество - отделяют друг от друга в делительной воронке, сушат над соответствующим осушителем. В качестве осушителей используют, как правило, безводные неорганические соли. Образующие с водой кристаллогидраты (сульфат натрия, хлорид кальция, перхлорат магния и др.).
Небольшие количества вещества можно перегонять, не пользуясь паровиком, а добавляя некоторое количество воды в перегонную колбу.
ЭКСТРАКЦИЯ. Для работы необходимы: делительная воронка на 100 мл, мерный цилиндр на 50 мл, четыре колбочки по 100 мл (рис. 1), колба на 250-300 мл, бюретка на 50 мл, пипетка на 20 мл, 0,4-0,5 моль/л раствор уксусной кислоты в изоамиловом спирте, 0,1н раствор щелочи, фенолфталеин.
Перед проведением опытов необходимо при помощи воды проверить на герметичность кран и стеклянную пробку делительной воронки. Изоамиловый спирт в количестве 40 мл с растворенной уксусной кислотой вливают в делительную воронку, добавляют 40 мл воды, насыщенной изоамиловым спиртом и проводят экстракцию.
Воду, насыщенную изоамиловым спиртом, получают, смешивая воду и изоамиловый спирт без уксусной кислоты и проводя все нижеописанные операции 1-3. Экстрагирование проводят 4 – 5 раз. Водный слой после каждого экстрагирования объединяют с предыдущими порциями, сливая его в колбу на 250-300 мл.
Последовательность операций при выполнении экстракции1.Закрыв делительную воронку стеклянной пришлифованной пробкой, правой рукой берутся за горлышко с пробкой, а левой – за кран так, чтобы суженная часть конуса помещалась в ладони, а пальцем можно было бы свободно поворачивать кран. Если держать в ладони сам корпус делительной воронки, то тепло руки повысит давление паров растворителя в воронке, в результате чего пробка и кран могут выскочить.
2.Делительную воронку поворачивают сливной трубкой кверху и осторожно приоткрывают кран. После сброса избыточного давления дают возможность жидкости, которая увлекается струей паров в сливную трубку, стечь обратно в воронку. Закрыв кран, воронку несколько раз встряхивают и снова открывают кран. Интенсивное встряхивание и выравнивание давления в делительной воронке с атмосферным давлением повторяют несколько раз для обеспечения достижения равновесия фаз.
3.Укрепив делительную воронку на штативе, ожидают разделения фаз. Открывают пробку и сливают нижнюю (водную) фазу в колбочку. Верхнюю фазу оставляют в воронке.
Затем в делительную воронку добавляют тот же (40 мл) объем воды, насыщенной изоамиловым спиртом, снова повторяют описанные выше операции 1-3. Экстрагирование проводят 4 – 5 раз. Из слитой каждый раз водной фазы берут пробы по 20 мл и титруют 0,1н раствором щелочи в присутствии фенолфталеина.
Результаты титрования записывают в табл. 1. По формуле (6) вычисляют К и делают выводы: остается ли К постоянным, независимым от концентрации уксусной кислоты; если нет, то почему. Сравнивают полученное значение константы распределения с литературными значениями. Обязательно указывают библиографические данные литературного источника.
Сщелочи = ![]() =
= ![]() =
=
Таблица
| i, номер экстрагирования | Объем, мл |
| К | |
| Аликвоты водной фазы | щелочи | |||
| 1. | ||||
Похожие работы
... -либо внешнего воздействия, и, наоборот, иногда требуются интенсивные внешние воздействия (катализаторы) для того, чтобы вызвать обратимые переходы изомеров друг в друга. Распространенное в органической химии явление, заключающееся в существовании двух или нескольких изомерных форм молекул, находящихся в состоянии динамического равновесия, называется таутомерией. В настоящее время установлено, ...
... деятельность. Поиск методов и форм обучения, способствующих воспитанию творческой личности, привел к появлению некоторых специфических способов обучения, одним из которых являются игровые методы. Реализация игровых методов обучения при изучении химии в условиях соблюдения дидактических и психолого-педагогических особенностей, повышает уровень подготовки учащихся. Слово «игра» в русском языке ...
... на новые программы и учебники этот вопрос становится наиболее острым. Наша школа перешла на новое учебники О.С. Габриеляна и новую программу, как и большинство школ Заволжского района, поэтому мы представляем календарно-тематическое планирование к курсу "Органическая химия" 10 класс. Тематическое планирование составлено согласно программе разработанной Департамента образовательных программ и ...
... . Работа предназначена учителям химии, а также может быть полезна студентам педагогических вузов и колледжей. 2.2.ПОЯСНИТЕЛЬНАЯ ЗАПИСКА Необходимость разработки элективного курса для учащихся 10-х классов «Решение задач по органической химии повышенного уровня сложности» обусловлена несколькими причинами. В соответствии с базисным учебным планом полной средней школы на изучение химии за 2 ...
0 комментариев