Навигация
Мл 0,001 Н розчину срібла нітрату відповідає 0,1660 мг KI
64717
знаков
14
таблиц
3
изображения
1 мл 0,001 Н розчину срібла нітрату відповідає 0,1660 мг KI.
Аргентометричне титрування йодидів можна проводити із застосуванням зовнішнього індикатора - нітрозо-крахмального паперу.
3.2.2. Іодкрахмальний метод КольтгофаЙодкрахмальний метод Кольтгофа використовують для визначення йодидів у присутності хлоридів і бромідів. В реакційну суміш, що містить йодиди, додають 1 краплю 0,001 Н розчину калія йодата, розчин крохмалю і розчин сірчаної кислоти розведеної. В результаті реакції утворюється йод, який забарвлює крохмаль в синій колір:
5I- + IO3- + 6Н+ = 3I2 + 3Н2O
Забарвлення йодкрахмального комплексу стійке лише у присутності йодид-йонів. Якщо їх зв'язати срібла нітратом, синє забарвлення розчину зникає. На різкість зміни забарвлення впливає кількість доданої води. У експрес-аналізі об'єм розчину повинен бути близько 15 мл. Метод дає гарні результати при титруванні йодидів у присутності хлоридів, якщо концентрація останніх не перевищує концентрацію йодидів більше ніж в 3 рази.
Метод також зручний при визначенні йодидів у присутності алкалоїдів пуринового ряду, барбітуратів та інших. Проте його не можна застосовувати для аналізу лікарських речовин, з якими може взаємодіяти йод (кислота аскорбінова, натрію саліцилат і інші).
У присутності бромідів титрування ускладнене, а перехід забарвлення від синього через сіре до жовтого наступає нерізко. Тому до додавання до розчину кислоти сірчаної, до реакційної суміші підливають 5 мл 10% розчину амонія карбонату.
До 10 мл розчину додають 1 краплю 0,001 Н розчину калія йодата, 2 мл свіжоприготованого розчину крохмалю і по краплях сірчану кислоту розведену до появи синього забарвлення рідини. Далі титрують 0,001 Н розчином срібла нітрату до переходу синього забарвлення в жовте.
1 мл 0,001 Н розчину срібла нітрату відповідає 0,166 мг KI.
3.2.3. Метод йодатометрії
Йодід-іони окислюються калія йодатом в середовищі кислоти хлороводневої спочатку до йоду:
5KI + KIO3 + 6НCL = 3I2 + 3Н2O + 6KCL
2I+5 + 10ē = I20 │ 1
2I- - 2ē = I20 │ 5
Йод, що виділився, титрують до йодмонохлорида:
2I2 + KIO3 + 6НCL = ICL + 3Н2O + KCL
I+5 + 4ē = I+ │ 1
I0 - 1ē = I+ │ 4
При розрахунку z = 3.
До 10 мл розчину додають 5 мл 25% розчину кислоти хлороводневої, 1 мл розчину крохмалю розчинного і швидко титрують 0,001 Н розчином калія йодата до появи яскраво-бурого забарвлення. Потім титрування проводять поволі, по краплях, до переходу забарвлення в лимонно-жовте.
1 мл 0,001 Н розчину калія йодата відповідає 55,33 мкг KI.
3.2.4. Метод перманганатометріїЙодид-іони окислюються калія перманганатом в середовищі кислоти хлороводневої розведеної:
2KMnO4 + 5KI + 16НCL = 5ICL + 7КCL + 2MnCL2 + 8Н2O
Mn+7 + 5ē = Mn+2 │ 2
I- - 2ē = I+ │ 5
При розрахунку z = 2.
Визначенню йодидів не заважає присутність в лікарських сумішах інших галогенідів, сульфатів, фосфатів, рибофлавіну, тіаміну броміду, глутамінової кислоти, теофіліну, еуфиліну, кофеїн-бензоата натрію, ефедрину гідрохлорида.
Заважають титруванню сильні відновники: кислота аскорбінова, натрію тіосульфат, цистеїн, ароматичні аміни, феноли і їх похідні і інші лікарські речовини, які можуть реагувати з калія перманганатом або йодмонохлоридом, що утворюються при титруванні .
До 10 мл розчину додають 5 мл 25% розчину кислоти хлороводневої і титрують 0,001 Н розчином калія перманганату до появи яскраво-бурого забарвлення розчину. Потім додають 1 мл розчину крохмалю розчинного і знову титрують по краплях до переходу забарвлення в лимонно-жовте.
1 мл 0,001 Н розчину калія перманганату відповідає 83,005 мкг KI.
3.2.5. Метод тіосульфатометріїЙодид-іони окислюються калію дихроматом або пергідролем в середовищі кислоти сірчаної розведеної:
K2Cr2O7 + 6KI + 7Н2SO4 = 3I2 + Cr2(SO4)3 + 4K2SO4 + 7Н2O
2Cr+6 + 6ē = 2Cr+3 │ 1
2I- - 2ē = I20 │ 3
H2O2 + 2KI + Н2SO4 = I2 + K2SO4 + 2Н2O
2O-1 + 2ē = 2O-2 │ 1
2I- - 2ē = I20 │ 1
Йод, що виділився, титрують натрію тіосульфатом до йодиду:
I2 + 2Na2S2O3 = 2NaI + Na2S4O6
4S+2 - 2ē = 4S+2.5 │ 1
2I0 + 2ē = 2I- │ 1
До 10 мл розчину додають 10 мл розчину кислоти сірчаної розведенної та 10 мл 0,001 Н розчину калію дихромату.
Йод, що виділився титрують 0,001 Н розчином натрію тіосульфату в присутності розчину крохмалю до переходу забарвлення в лимонно-жовте.
1 мл 0,001 Н розчину натрію тіосульфату відповідає 0,166 мг KI.
3.3. Вибір методу аналізу індивідуальної хімічної речовини KI 3.3.1. Мета роботи1. Використання доступного методу – титрування.
2. Зменшеня кількості препарату для аналізу – економічна доцільність.
3.3.2. Підготовка проби для аналізуДля досліджень використовувалась хімічно чиста речовина KI з вмістом KI – 99,2%.
Випробування методики проводилися на контрольному розчині, що був приготовлений таким чином: наважка масою 54 мг сухої речовини KI була розчинена у 100 мл дистильованої води. Таким чином кількість KI в 0,5 мл приговленого розчину еквівалентна кількості KI в одній таблетці з маркуванням 200 (261 мкг KI на 1 таблетку), та також двом таблеткам з маркуванням 100 (130 мкг KI на 1 таблетку).
Оскільки з економічної доцільності для аналізу потрібно брати не більше двох таблеток з маркуванням 200, то і контрольний розчин брали виходячи з такої самої кількості KI, а саме 1 мл.
3.3.3. Метод ФаянсаПри виконанні аналізу за методом Фаянса відбувається наступна реакція:
KI + AgNO3 = AgI¯ + KNO3
В одній пробі об’ємом 1 мл міститься 540 мкг KI.
Згідно закону еквівалентів розраховуємо кількість еквівалентів KI:
m(KI) = 540*10-6 г
me(KI) = 166 г/моль
ne(KI) = m(KI) / me(KI) = 540*10-6 / 166 = 3,25*10-6екв.
Відповідно осаду AgI випадає:
ne(AgI) = ne(KI) = 3,25*10-6 екв.
me(AgI) = 235 г/моль
m(AgI) = ne(AgI) * me(AgI) = 3,25*10-6 * 235 = 736*10-6 г = 0,736 мг
Таку малу кількість осаду майже неможливо розгледіти, тим паче побачити зміну забарвлення еозината натрію на поверхні осаду.
В зв'язку з цим метод Фаянса був визнаний неприйнятним.
3.3.4. Метод КольтгофаЗгідно з методикою до розчину додають 1 краплину (0,1 мл) KIO3 і відбувається наступна реакція:
5KI + KIO3 + 3Н2SO4= 3I2 + 3Н2O + 3K2SO4
Крохмаль повинен прореагувати з йодом та забарвитись в синій колір. Розрахуємо кількість йоду, що має виділитися:
Сн(KIO3) = 1*10-3 моль/л
V(KIO3)= 0,03 мл = 3*10-5 л
ne(KIO3) = Сн(KIO3) * V(KIO3) = 1*10-3 * 3*10-5 = 3*10-8 екв.
ne(I2) = ne(KIO3) = 3*10-8 екв.
Vр-н = 12 мл = 12*10-3 л (10 мл проби + 2 мл крохмалю)
ne(I2) = Vр-н * Сн(I2)
Сн(I2) = ne(I2) / Vр-н = 3*10-8 / 12*10-3 = 2,5*10-6 моль/л
Така концентрація знаходиться за межею чутливості крохмального індикатора, а отже поява синього забарвлення не відбудеться.
В зв’язку з цим метод Кольтгофа також був визнаний неприйнятним.
3.3.5. Метод тіосульфатометріїВ основі даного методу лежать реакції окиснення йодиду з утвореням йоду окисниками (калію дихромат або пергідроль) згідно рівнянь:
K2Cr2O7 + 6KI + 7Н2SO4 = 3I2 + Cr2(SO4)3 + 4K2SO4 + 7Н2O
H2O2 + 2KI + Н2SO4 = I2 + K2SO4 + 2Н2O
Потім йод титрують тіосульфатом натрію. Реакція проходить за наступним рівнянням:
I2 + 2Na2S2O3 = 2NaI + Na2S4O6
Отже ми бачимо, що в розчині знову утворюється йодид, який одразу ж реагує з надлишком окисника і знову утворюється йод. Таким чином реакція перебігає по такому колу до тих пір, доти не прореагує весь надлишок окисника.
В зв’язку з цим метод тіосульфатометрії також був визнаний неприйнятним.
3.3.6. Метод йодатометріїПід час проведеня аналізу відбувається така реакція:
5KI + KIO3 + 6НCL = 3I2 + 3Н2O + 6KCL
Виділений йод викликає синє забарвлення крохмального індикатора. Проведемо розрахунки.
Оскільки в одній пробі об’ємом 1 мл міститься 540 мкг KI, то маємо такі результати:
m(KI) = 540*10-6 г
me(KI) = 166 г/моль
ne(KI) = m(KI) / me(KI) = 540*10-6 / 166 = 3,25*10-6екв.
ne(I2) = ne(KIO3) = 3,25*10-6 екв.
Vр-н = 16 мл = 16*10-3 л (10 мл проби + 5 мл 25% HCL + 1 мл крохмалю)
ne(I2) = Vр-н * Сн(I2)
Сн(I2) = ne(I2) / Vр-н = 3,25*10-6 / 16*10-3 = 2,03*10-4 моль/л
Отже ми бачимо досить високу концентрацію йоду та відповідно інтенсивне синє забарвлення індикатора.
Після того, як весь йодид перетворюється в йод, починається друга стадія реакції. Йод реагує з йодатом з утвореням йодмонохлориду:
2I2 + KIO3 + 6НCL = ICL + 3Н2O + KCL
Поступово, по мірі зменшеня концентрації йоду, забарвлення з синього переходить в буре, а в момент точки еквівалентності, коли йод прореагує повністю, переходить в лимонно жовте.
Даний метод повністю задовольнив всім вимогам і дозволив отримати результати титрування (Таблиця 1).
Таблиця 1.
| № п/п | Об’єм алікв-оти, мл | Об’єм титранту, мл | Середнє значення об’єму титранту, мл | Маса KI в аліквоті, експерементальне значення, мкг | Маса наважки, мкг | Маса KI в нава-жці, істине значе-ня, мкг | Вміст, % | Відно-сна поми-лка,% |
| 1 | 1,00 | 9,20 | 9,16 | 506,85 | 540,00 | 535,68 | 94,62 | 5,38 |
| 2 | 1,00 | 9,20 | ||||||
| 3 | 1,00 | 9,10 |
CH(KIO3) = 0,001 моль/л
V(KIO3) = 9,16 мл
m(KI) = CH(KIO3) * V(KIO3) * me(KI) =
= 1*10-3 * 9,16*10-3 * 55,3333 = 506,85 *10-6 г = 506,85 мкг
Маса KI в наважці = 540 мкг * 99,2% / 100 = 535,68 мкг
Вміст = mалікв / mнаважк * 100 = 506,85 / 535,68 * 100 = 93,86 %
Відносна помилка = (mнаважк - mалікв) / mнаважк * 100 =
= (535,68 - 506,85) / 535,68 * 100 = 6,14 %
3.3.7. Метод перманганатометріїПід час проведення аналізу відбувається така реакція:
2KMnO4 + 5KI + 16НCL = 5ICL + 7КCL + 2MnCL2 + 8Н2O
Кінцевим продуктом є йодмонохлорид. Виділений йод викликає синє забарвлення крохмального індикатора, як і в методі йодатометрії.
При поступовому додаванні калію перманганату, забарвлення з синього переходить в буре, а в момент точки еквівалентності, коли йод прореагує повністю, переходить в лимонно жовте.
Даний метод повністю задовольнив всім вимогам і дозволив отримати результати титрування (Таблиця 2).
Таблиця 2.
| № п/п | Об’єм алікв-оти, мл | Об’єм титран-ту, мл | Серед-нє значе-ня об’єму титран-ту, мл | Маса KI в аліквоті, експеремен-тальне значення, мкг | Маса наважки, мкг | Маса KI в нава-жці, істине значе-ня, мкг | Вміст, % | Відносна помилка, % |
| 1 | 1,00 | 6,20 | 6,16 | 511,28 | 540,00 | 535,68 | 95,45 | 4,55 |
| 2 | 1,00 | 6,20 | ||||||
| 3 | 1,00 | 6,10 |
CH(KMnO4) = 0,001 моль/л
V(KMnO4) = 6,16 мл
m(KI) = CH(KMnO4) * V(KMnO4) * me(KI) =
= 1*10-3 * 6,16*10-3 * 83 = 511,28 *10-6 г = 511,28 мкг
Маса KI в наважці = 540 мкг * 99,2% / 100 = 535,68 мкг
Вміст = mалікв / mнаважк * 100 = 511,28 / 535,68 * 100 = 95,45 %
Відносна помилка = (mнаважк - mалікв) / mнаважк * 100 =
= (535,68 - 511,28) / 535,68 * 100 = 4,55 %
ВисновокОтже методи перманганатометрії та йодатометрії дають відтворювані і близькі один до одного результати (Таблиця 3).
Таблиця 3.
| Метод | Маса KI, мкг | Вміст KI, % | Відносна помилка, % | ||
| Експерем-ентальне значеня | Істине значеня | Експерем-ентальне значеня | Істине значеня | ||
| Йодато-метрія | 506,85 | 535,68 | 94,62 | 99,20 | 5,38 |
| Перманган-атометрія | 511,28 | 535,68 | 95,45 | 99,20 | 4,55 |
Використання обох цих методів прийнятне для аналізу розчинів, що містять KI.
3.4. Вибір оптимальних умов пробо підготовкиОсновною операцією пробопідготовки лікарського препарату є екстракція.
3.4.1. Екстракція водоюПершу екстракцію проведимо дистильованною водою за схемою:
5 таблеток розтирають у ступці. Змішують з 8 мл води, збовтують протягом 15 хв. (механічно) у центрифужной пробірці місткістю 10 мл, закритою пластиковою пробкою. Потім центрифугують протягом 10 хв.
Розчин зливають, знову додають 8 мл. води та другий раз збовтують протягом 15 хв. І знову центрифугують.
Потім знову зливають розчин і втретє повторюють збовтування та центрифугування.
Отриманий розчин доводять до 50 мл і титрують аліквотами по 10 мл.
За результатами титрування отримані такі результати (Таблиця 4)
Таблиця 4.
| № п/п | Об’єм алікв-оти, мл | Об’єм титран-ту, мл | Середнє значеня об’єму титран-ту, мл | Маса KI в алікв-оті, мкг | Маса KI в таблетці, мкг | Вміст KI, % | Відносна помилка, % | |
| експерем-ентальне значеня | Істине значеня | |||||||
| 1 | 10,00 | 2,80 | 2,78 | 767,75 | 153,55 | 260,00 | 59,06 | 40,94 |
| 2 | 10,00 | 2,70 | ||||||
| 3 | 10,00 | 2,90 | ||||||
| 4 | 10,00 | 2,70 | ||||||
CH(KIO3) = 0,001 моль/л
V(KIO3) = 2,775 мл
CH(KIO3) * V(KIO3) * me(KI) * Vпроби
m(KI) = ---------------------------------------------------------- =
Vаліквоти
= 1*10-3 * 2,775*10-3 * 55,3333 * 50 / 10 = 567,75 *10-6 г = 767,75 мкг
Вміст = mалікв / mнаважк * 100 = 767,75 / (260 * 5) * 100 = 59,06 %
Відносна помилка = (mнаважк - mалікв) / mнаважк * 100 =
= (260 - 153,55) / 260 * 100 = 40,94 %
Отже ми бачимо, що екстракція склала лише 59 %.
Причиною цього можуть бути органічні складові таблетки, які зв’язують калію йодид і знижують його розчинність.
В зв’язку з цим було вирішено випробувати інші методики екстракції, в яких до розчинника було додано азотну кислоту.
3.4.2. Екстракція 2 % розчином HNO3В другій спробі екстракції була використана досить висока концентрація HNO3.
У 100 мл води було розчинено 2 мл HNO3 концентрованої. Відповідно був отриманий 2 % розчин HNO3.
Під час екстракції таблетка розчинилась повністю за декілька хвилин, але отримані результати виявились дещо заниженими (Таблиця 5).
Таблиця 5.
| № п/п | Об’єм алікв-оти, мл | Об’єм титран-ту, мл | Середнє значеня об’єму титран-ту, мл | Маса KI в алікв-оті, мкг | Маса KI в таблетці, мкг | Вміст KI, % | Відносна помилка, % | |
| експерем-ентальне значеня | Істине значеня | |||||||
| 1 | 10,00 | 3,20 | 3,25 | 899,16 | 179,83 | 260,00 | 69,17 | 30,83 |
| 2 | 10,00 | 3,30 | ||||||
| 3 | 10,00 | 3,30 | ||||||
| 4 | 10,00 | 3,20 | ||||||
CH(KIO3) = 0,001 моль/л
V(KIO3) = 3,25 мл
CH(KIO3) * V(KIO3) * me(KI) * Vпроби
m(KI) = ---------------------------------------------------------- =
Vаліквоти
= 1*10-3 * 3,25*10-3 * 55,3333 * 50 / 10 = 899,16 *10-6 г = 899,16 мкг
Вміст = mалікв / mнаважк * 100 = 899,16 / (260 * 5) * 100 = 69,17 %
Відносна помилка = (mнаважк - mалікв) / mнаважк * 100 =
= (260 - 179,83) / 260 * 100 = 30,83 %
Отже ми бачимо, що азотна кислота підвищила розчинність калію йодиду, але будучи сильним окисником прореагувала з частиною йодиду і тим самим знизила результати титрування.
3.4.3. Екстракція 0,02 % розчином HNO3В зв’язку з цим в третій спробі екстракції була знижена концентрація HNO3.
У 500 мл води було розчинено 0,1 мл HNO3 концентрованої. Відповідно був отриманий 0,02 % розчин HNO3.
Під час екстракції таблетка також розчинилась повністю за десять хвилин, але у цій спробі отримані результати виявились прийнятними (Таблиця 6).
Таблиця 6.
| № п/п | Об’єм алікв-оти, мл | Об’єм титран-ту, мл | Середнє значеня об’єму титран-ту, мл | Маса KI в алікв-оті, мкг | Маса KI в таблетці, мкг | Вміст KI, % | Відносна помилка,% | |
| експерем-ентальне значеня | Істине значеня | |||||||
| 1 | 10,00 | 7,10 | 7,10 | 1178,60 | 235,72 | 260,00 | 90,66 | 9,34 |
| 2 | 10,00 | 7,20 | ||||||
| 3 | 10,00 | 7,00 | ||||||
CH(KIO3) = 0,001 моль/л
V(KIO3) = 7,1 мл
CH(KIO3) * V(KIO3) * me(KI) * Vпроби
m(KI) = ---------------------------------------------------------- =
Vаліквоти
= 1*10-3 * 7,1*10-3 * 55,3333 * 30 / 10 = 1178,6 *10-6 г = 1178,6 мкг
Вміст = mалікв / mнаважк * 100 = 1178,6 / (260 * 5) * 100 = 90,66 %
Відносна помилка = (mнаважк - mалікв) / mнаважк * 100 =
= (260 - 235,72) / 260 * 100 = 9,34 %
Для перевірки результатів такої методики екстракції, було проведено ще одне титрування методом перманганатометрії. Результати титрування були такі (Таблиця 7).
Таблиця 7.
| № п/п | Об’єм алікв-оти, мл | Об’єм титран-ту, мл | Середнє значеня об’єму титран-ту, мл | Маса KI в алікв-оті, мкг | Маса KI в таблетці, мкг | Вміст KI, % | Відносна помилка,% | |
| експерем-ентальне значеня | Істине значеня | |||||||
| 1 | 10,00 | 4,80 | 4,76 | 1185,24 | 237,05 | 260,00 | 91,17 | 8,83 |
| 2 | 10,00 | 4,80 | ||||||
| 3 | 10,00 | 4,70 | ||||||
CH(KMnO4) = 0,001 моль/л
V(KMnO4) = 4,76 мл
CH(KMnO4) * V(KMnO4) * me(KI) * Vпроби
m(KI) = ---------------------------------------------------------- =
Vаліквоти
= 1*10-3 * 4,76*10-3 * 83 * 30 / 10 = 1185,24 *10-6 г = 1185,24 мкг
% = mпракт / mтеор * 100 = 1185,24 / (260*5) * 100 = 91,17 %
Відносна помилка = (mнаважк - mалікв) / mнаважк * 100 =
= (260 - 237,048) / 260 * 100 = 8,83 %
ВисновокМожна побачити, що результати двох методів титрування співпадають, а отже вимірювання проведені правильно (Таблиця 8).
Таблиця 8.
| Метод аналізу, метод екстракції | Маса KI в аліквоті, мкг | Маса KI в таблетці, мкг | Вміст KI, % | Відносна помилка, % | |
| експерементальне значеня | Істине значеня | ||||
| Йодато-метрія, Н2О | 767,75 | 153,55 | 260,00 | 59,06 | 40,94 |
| Йодато-метрія, 2% HNO3 | 899,16 | 179,83 | 260,00 | 69,17 | 30,83 |
| Йодато-метрія, 0,02% HNO3 | 1178,60 | 235,72 | 260,00 | 90,66 | 9,30 |
| Перманган-атометрія, 0,02% HNO3 | 1185,24 | 237,05 | 260,00 | 91,17 | 8,83 |
Вище наведені дослідження показують, що екстракція є дуже важливою стадією в аналізі лікарських форм, яка значно впливає на результати досліджень.
Найбільш прийнятною виявилась екстракція 0,02 % розчином HNO3.
3.5. Встановлення чутливості методики аналізуТакож дуже важливо виявити чутливість методу, тобто мінімальну кількість KI, яку дозволяє визначити методика.
Найменша концентрація, яку фіксує крохмальний індикатор така:
Сн(I2) = 5*10-6 моль/л
Звідси розраховуємо:Сн(I2) = ne(I2) / Vр-н
ne(I2) = Сн(I2) * Vр-н = 5*10-6 * 12*10-3 = 6*10-8 моль/л
m(KI) = ne(I2) * me(KI) = 6*10-8 * 55,3333 = 3,3*10-6 г = 3,3 мкг
Але дослідження показали, що і набагато більші кількості KI не вдається виявити обраними методами, оскільки синє, а за ним і буре забарвлення дуже слабкі і точно зафіксувати перехід до жовтого неможливо. В зв’язку з цим було спробувано виявити чутливість реакції експериментальним шляхом. Результати цих випробувань наведені у таблиці 9.
Таблиця 9.
| № п/п | Об’єм аліквоти контрольного розчину, мл | Кількість KI | Встановленя кінцевої точки титрування | Вміст, % | Відносна помилка, % | |
| Маса у аліквоті, мкг | Кількість таблеток, шт | |||||
| 1 | 0,20 | 108,00 | 0,42 | Неможливо | --- | --- |
| 2 | 0,40 | 216,00 | 0,83 | Неможливо | --- | --- |
| 3 | 0,50 | 270,00 | 1,04 | Не точно | 60 - 80 | 20 - 40 |
| 4 | 0,60 | 324,00 | 1,25 | Задовільно | 90,66 | 9,30 |
| 5 | 1,00 | 540,00 | 2,08 | Задовільно | 91,17 | 8,83 |
Для точного та якісного виконання аналізу необхідно брати на одне титрування півтори – дві таблетки з маркуванням 200, та три – чотири таблетки з маркуванням 100.
Висновок
Хімічний елемент – Йод, відкритий у 1811 р. Бернаром Куртуа, у наш час знайшов широке застосування в промисловості, техніці, фотосправі та в медицині і не тільки як антисептичний засіб, а як мікроелемент, що дуже важливий для підтримки здоров'я щитовидної залози. Вивчення властивостей Йоду вже привело до появи біологічно активних добавок, що містять мікроелемент Йод.
У зв’язку з цим досить актуальним є контроль якості препаратів, що містять Йод. Згідно аналітичної нормативної документації, аналіз препаратів проводять методом потенціометрії з використанням срібного індикаторного електроду або з використанням рідинної хроматографії.
Оскільки вище зазначені методики є досить коштовними і потребують спеціального обладнання, була поставлена мета розробити більш дешеву та доступну методику контроля якості препаратів, що містять йод.
Підчас роботи були виконані наступні завдання:
1. Використання доступного методу – титрування.
2. Зменшеня кількості препарату для аналізу – економічна доцільність.
Для аналізу були обрані три препарати, які представлені на Українському фармацевтичному ринку:
1. Йодомарин – виробник “Берлін-Хемі”, Німеччина
2. Йодбаланс – виробник “Нікомед”, Норвегія
3. Йодид – виробник “Фармак”, Україна
Було випробувано п’ять методик, основою яких є титрування:
1. Аргентометрія за методом Фаянса
2. Йодкрахмальний метод Кольтгофа
3. Окисно-відновний метод з використанням йодатометрії
4. Окисно-відновний метод з використанням тіосульфатометрії
5. Окисно-відновний метод з використанням перманганатометрії
Дослідження показали, що осаджувальні методики титрування Фаянса і Кольтгофа не дають бажаних результатів, порівняно з окисно-відновними методиками.
В методі Фаянса виділяється дуже мала кількість осаду, яку майже неможливо розгледіти, тим паче побачити зміну забарвлення еозината натрію на поверхні осаду.
В методі Кольтгофа концентрація йоду знаходиться за межею чутливості крохмального індикатора, а отже поява синього забарвлення не відбувається.
Окисно-відновна методика тіосульфатометрії також не дала бажаних результатів, в зв’язку з тим, що в розчині при титруванні йоду знову утворюється йодид, який одразу ж реагує з надлишком окисника і знову утворюється йод. Таким чином реакція перебігає по такому колу до тих пір, доти не прореагує весь надлишок окисника.
Найбільш точними та ефективними виявились методики йодатометрії та перманганатометрії, які показали відтворювані результати. Результати знаходились в межах норми, яка вказана в аналітичній нормативній документації: 100 ± 15 %.
| Метод | Маса KI, мкг | Вміст KI, % | Відносна помилка,% | ||
| Експерем-ентальне значеня | Істине значеня | Експерем-ентальне значеня | Істине значеня | ||
| Йодато-метрія | 506,85 | 535,68 | 94,62 | 99,20 | 5,38 |
| Перманган-атометрія | 511,28 | 535,68 | 95,45 | 99,20 | 4,55 |
Серед цих двох методик краще вибрати перманганатометрію, оскільки за однакової точності вона є більш дешевою: (Калію перманганат дешевше, ніж калію йодат).
Також було показано, що екстракція є дуже важливою стадією в аналізі лікарських форм, яка значно впливає на результати досліджень.
Найбільш прийнятною виявилась екстракція 0,02 % розчином HNO3, яка дала результат екстракції – 91 %. Порівняно з нею екстракція водою дала значно менший результат – 59 %.
| Метод аналізу, метод екстракції | Маса KI в аліквоті, мкг | Маса KI в таблетці, мкг | Вміст KI, % | Відносна помилка, % | |
| експерементальне значеня | Істине значеня | ||||
| Йодато-метрія, Н2О | 767,75 | 153,55 | 260,00 | 59,06 | 40,94 |
| Йодато-метрія, 2% HNO3 | 899,16 | 179,83 | 260,00 | 69,17 | 30,83 |
| Йодато-метрія, 0,02% HNO3 | 1178,60 | 235,72 | 260,00 | 90,66 | 9,30 |
| Перманган-атометрія, 0,02% HNO3 | 1185,24 | 237,05 | 260,00 | 91,17 | 8,83 |
Для найбільш точного та якісного виконання аналізу необхідно брати на одне титрування півтори – дві таблетки з маркировкою 200, та три – чотири таблетки з маркировкою 100.
Менша кількість призводить до зниженя точності, або навіть неможливості аналізу. Більша кількість призводить до перевитрати таблеток, які коштують відносно дорого.
| № п/п | Об’єм аліквоти контрольного розчину, мл | Кількість KI | Встановленя кінцевої точки титрування | Вміст, % | Відносна помилка, % | |
| Маса у аліквоті, мкг | Кількість таблеток, шт | |||||
| 1 | 0,20 | 108,00 | 0,42 | Неможливо | --- | --- |
| 2 | 0,40 | 216,00 | 0,83 | Неможливо | --- | --- |
| 3 | 0,50 | 270,00 | 1,04 | Не точно | 60 - 80 | 20 - 40 |
| 4 | 0,60 | 324,00 | 1,25 | Задовільно | 90,66 | 9,30 |
| 5 | 1,00 | 540,00 | 2,08 | Задовільно | 91,17 | 8,83 |
Екстракцію 0,02 % розчином HNO3, разом з титруванням калію йодатом або калію перманганатом доцільно використовувати для перевірки якості препаратів, що містять йод, оскільки вони забезпечують достатню точність при набагато менших затратах, ніж методи вказані у аналітичній нормативній документації.
Список використаної літератури
1. Популярная библиотека химических элементов. – М.: Наука 1973г.
2. Энциклопедический словарь юного химика. Сост. В.А.Крицман, В.В.Станцо. – М.: Педагогика, 1982г.
3. Справочник по химии для поступающих в ВУЗы. Под редакцией А.Т.Пилипенко. – Киев: Наукова думка, 1971г.
4. Ермолаев М.В. Биологическая химия. – М.: Медицина, 1983г.
5. Загальна та неорганічна хімія. Під редакцією Є.Я.Лєвітіна. – Вінніця: Нова книга, 2003р.
6. Селезнев К.А. Аналитическая химия. – М.: Высшая школа, 1963г.
7. Гайдукевич А.Н. Аналітична хімія. – Х.: Основа, 2000г.
8. Государственная Фармакопея СССР 10-е изд. - М.: Медицина, 1968г.
9. Международная Фармакопея, Женева. - М.: Медицина, 1981г.
10. Гиллебранд В.Ф. Практическое руководство по неорганическому анализу – М.: Химия, 1966г.
11. Кольтгоф И.М., Стенгер В.А. Объёмный анализ. т.1,2 – М.: Госхимиздат, 1950г.
12. Арзамасцев А.Р. Фармацевтическая химия. – М.: Гэотар-Медиа, 2006г.
13. Аналітична нормативна документація на препарат “Йодид” – “Нікомед” від 21/05/2003 № 228.
14. Аналітична нормативна документація на препарат “Йодомарин” – “Берлін-Хемі АГ” від 16/12/2002 № 470.
15. Определение массовой доли йода в пищевых продуктах и сырье титриметрическим методом. Методические указания мук 4.1.1106-02. Методика Министерства Здравоохранения РФ от 14/02/2002
Похожие работы
... повинна вписуватися в технологічну схему як один з елементів процесу, інтенсифікуючи або, принаймні, не зменшуючи продуктивності праці. 2. Види внутрішньоаптечного контролю й основні форми його удосконалення 2.1 Контроль якості ліків в умовах аптек У державному нормуванні виробництва лікарських препаратів велика увага приділяється контролю якості готового продукту. Якість лікарського ...

... амілази і пуллуланази або альфа-амілази і глюкоамілази, активних при високій температурі. Однак складність культивування анаеробних бактерій в заводських умовах робить цей спосіб отримання препарату амілолітичних ферментів практично непридатним. З літературних даних відомо, що найбільш термостабільними гідролітичними ферментами, такими як альфа-амілази, пуллуланази, а в ряді випадків і протеази є ...
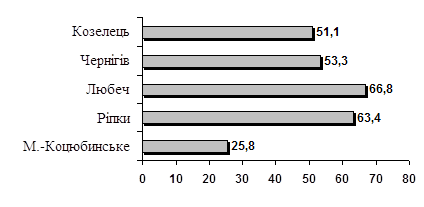
... Ін-т ендокрин. та обміну речовин ім. В.П. Комісаренка АМНУ. – №2003077197; заявл. 30.07.03; опубл. 15.06.2004, Бюл. №6. АНОТАЦІЯ Турчин В.І. Йодний дефіцит та патологія щитоподібної залози серед дитячого населення у Північному регіоні України. – Рукопис. Дисертація на здобуття наукового ступеня кандидата медичних наук за спеціальністю 14.01.14 – ендокринологія. – Інститут ендокринології та ...
... + 6e + 14H = 2Cr + 7H2O Робочі розчини K2Cr2O7 готують по точній наважці без використання встановлюючи речовин. Розчини дихромату стабільні і довго зберігаються без змін. Аналіз поводять методом прямого титрування в присутності індикатора дифениламин, забарвлений в синій колір при найменшому надлишку дихромату. Титанометрія – цей метод оснований на відновленні аналізуючої речовини , при цьому ...
0 комментариев