Навигация
5.3 Контроль качества
Контроль качества - это часть “Правил...”, посвященная отбору проб, проведению испытаний и выдаче соответствующих документов, гарантирующих, что все необходимые испытания действительно были проведены; что в производстве были использованы сырье, вспомогательные, упаковочные и маркировочные материалы требуемого качества и что готовый продукт был реализован только в том случае, если его качество отвечало требованиям соответствующей нормативной документации.
5.3.1 Каждое фармацевтическое предприятие должно иметь отдел контроля качества (ОКК). ОКК является самостоятельным и независимым структурным подразделением фармацевтического предприятия и возглавляется квалифицированным специалистом с достаточным стажем работы. В своей работе ОКК руководствуется государственными и отраслевыми документами, регламентирующими его деятельность.
5.3.2 Система контроля качества (объекты контроля, контрольные операции и их последовательность, техническое оснащение, методы, средства механизации, автоматизации и компьютеризации контрольных операций) являются неотъемлемой частью производственного процесса.
5.3.3 Основными требованиями, предъявляемыми к ОКК, являются следующие:
-наличие высококвалифицированного персонала; оснащение полным набором необходимого современного лабораторного оборудования, контрольно-измерительных приборов и реактивов; наличие необходимой утвержденной нормативной документации, а также аналитических методик и/или инструкций по проведению постадийного контроля процесса производства;
-проведение отбора проб (сотрудниками ОКК или в их присутствии) исходного сырья, вспомогательных, упаковочных и маркировочных материалов, полупродуктов и готового продукта в соответствии с утвержденными инструкциями;
-осуществление входного контроля исходного сырья, вспомогательных, упаковочных и маркировочных материалов, полупродуктов и готового продукта по соответствующей нормативной документации;
-осуществление контроля за соответствием их установленным требованиям при передаче из помещений для хранения в производство и из цеха в цех и/или на склад;
-валидация методов проведения анализов;
-осуществление контроля качества готового продукта и наблюдение за стабильностью препаратов при хранении в течение одного года после окончания установленных сроков годности, но не менее трех лет;
-участие в планировании, организации и проведении постадийного контроля процесса производства (совместно с работниками цеховых лабораторий, цехов и/или отделов);
-регистрация всех проведенных во время изготовления серии готового лекарственного средства анализов и полученных результатов, в том числе результатов проведения постадийного контроля процесса производства. Любое отклонение должно быть зарегистрировано и тщательно проанализировано;
-хранение достаточного количества образцов исходного сырья, вспомогательных, упаковочных и маркировочных материалов, лекарственных веществ и готовых лекарственных средств для обеспечения возможности проверки ОКК или органами государственного контроля во время хранения. Образцы каждой серии готового продукта в окончательной упаковке должны храниться в рекомендованных условиях в течение одного года после окончания срока годности готового продукта, но не менее трех лет. Образцы активных исходных веществ должны храниться в течение одного года после истечения срока годности лекарственного средства, в состав которого они входят, но не менее трех лет. Вспомогательные вещества (кроме растворителей, газов и воды) должны храниться минимально три года;
-хранение паспортов на все изготовленные серии лекарственных веществ или готовых лекарственных средств; копии результатов анализов исходного сырья, вспомогательных, упаковочных и маркировочных материалов, лекарственных веществ, готовых лекарственных средств и постадийного контроля процесса производства в течение одного года после окончания срока годности готового продукта, но не менее трех лет.
6. Регистрация данных о качестве
Зарегистрированные данные о качестве должны содержать сведения, удостоверяющие непосредственно или косвенно, удовлетворяет ли продукция техническим требованиям.
Зарегистрированные данные о качестве продукции должны обеспечивать свидетельство того, что элементы системы качества, подпадающие под требования ИСО 9001, ИСО 9002 или ИСО 9003, внедрены.
Если результаты не будут убедительно продемонстрированы, в зарегистрированных данных о качестве следует указать, что сделано для того, чтобы исправить положение.
Предприятие должно подготавливать, вести, хранить в безопасном месте и защищать от несанкционированного доступа и изменений зарегистрированные данные о качестве.
Данные должны быть легко доступны. Их можно хранить или копировать в любой подходящей форме, например, на бумажном или электронном носителе.
Копии должны содержать всю релевантную информацию, которая имеется в оригинальных данных.
Предприятие должно предусмотреть способ перевода требований контракта в требования, касающиеся представления, сохранения и утилизации регистрационных данных о качестве.
Предприятие должно предусмотреть возможность предоставления покупателю нужных ему документов.
7. Внутренняя проверка качества
Внутренняя проверка качества, требуемая согласно ИСО 9001 и ИСО 9002, должна проводиться предприятием, чтобы определить, являются ли различные элементы системы качества эффективными и пригодными для достижения установленных целей качества.
В плане внутренних проверок следует определить их периодичность.
Предприятие должно отобрать и назначить компетентных ревизоров с учетом критериев отбора.
Периодические внутренние проверки выполняются, чтобы:
- определить адекватность и соответствие элементов системы качества требованиям к документации и реализации;
- определить эффективность реализованной системы качества в достижении заданных целей качества;
- определить соответствие регламентирующим требованиям;
- обеспечить возможность усовершенствования систему качества предприятия;
- способствовать проверкам качества сторонней организацией.
8. Контроль процесса производства
С целью предотвращения выпуска готового продукта, не соответствующего требованиям нормативной документации, должен проводиться постадийный контроль процесса производства, который осуществляется сотрудниками цеховой лаборатории (регулярно) и отдела контроля качества (периодически) в соответствии с действующими отраслевыми документами, технологическими регламентами и письменными инструкциями. Периодичность проверок определяется руководством предприятия и отдела контроля качества применительно к данному продукту и процессу производства.
В ходе постадийного контроля проверяется:
-соответствие используемых сырья, вспомогательных, упаковочных и маркировочных материалов и полупродуктов требованиям нормативной документации;
-санитарное состояние цехов, рабочих мест и оборудования;
-выполнение регламентированных технологических операций и соблюдение технологических режимов работы.
Результаты постадийного контроля отражаются в соответствующих журналах и в досье на препарат. При обнаружении отклонений от режимов и норм технологического процесса должны быть выявлены причины и приняты меры по их ликвидации, что должно быть документально оформлено и внесено в досье.
Библиография
1. РД 64-125-91 “Правила организации производства и контроля качества лекарственных средств (GMP)”.М., Минмедпром СССР, 1991.
2. МУ 42-51-1-93 - МУ 42-51-26-93 “Организация и контроль производства лекарственных средств. Стерильные лекарственные средства” М., МЗ РФ, 1993.
3. ОСТ 42-504-96 “Контроль качества лекарственных средств на промышленных предприятиях и в организациях. Основные положения”.
4. Положение об отделе технического контроля предприятия, комбината, производственного, научно-производственного объединения Минмедбиопрома СССР (Утверждено приказом Минмедбиопрома СССР от 17.12.86 г. № 967).
5. Шилова С.В., Пузакова С.М., Назаров А.Д., Никульшина Н.И., Граковская Л.К. “Организация производства лекарственных средств с учетом правил GMP”. Хим.-фарм. производство. Обзорная информация, вып.2, М., ВНИИСЭНТИЮ, Минмедпром СССР, 1990, с.1-26
6. Граковская Л.К., Шилова С.В., Пузакова С.М., Мотина Г.Л., Плетень А.П. “Значение правил GMP для развития фармацевтической промышленности России”. Ж. Технология чистоты., М., 1993, № 2, с.12-14.
Похожие работы
... и контролирует правильность оформления прописей для индивидуального производства лекарств. Ведет текущую и отчетную документацию. Проводит целевое фармацевтическое исследование аптек, других учреждений с целью оценки состояния контроля качества лекарственных средств при их изготовлении, транспортировке, хранении и отпуске. Экспресс-анализ лекарственных форм. Необходимость внутриаптечного ...
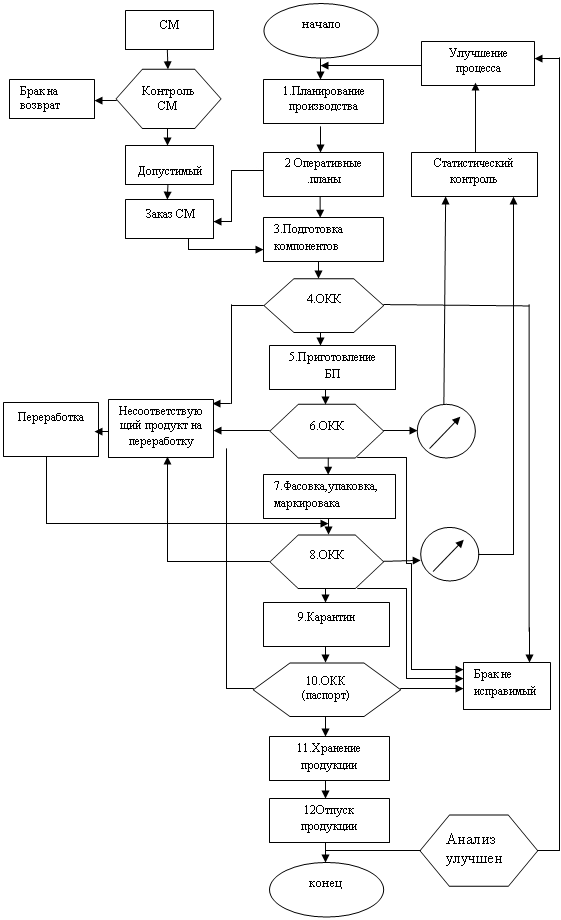
... настоящего стандарта обязательны для исполнения всеми участками процесса. Требования настоящего СТО распространяются на действия по производству лекарственного препарата Нитокс 200 и область возможного его улучшения. Сознательное нарушение требований настоящего стандарта организации является нарушением исполнительской дисциплины и может быть основанием для административного взыскания. 2. ...
... показателей и методов контроля качества Л С. Он должен обеспечивать разработку эффективного и безопасного Л С. Новый ОСТ предусматривает наличие двух категорий стандартов качества: I. Государственные стандарты качества лекарственных средств (ГСКЛС), к которым относятся: общая фармакопейная статья (ОФС) и фармакопейная статья (ФС); II. Стандарт качества (СКЛС); фармакопейная ...

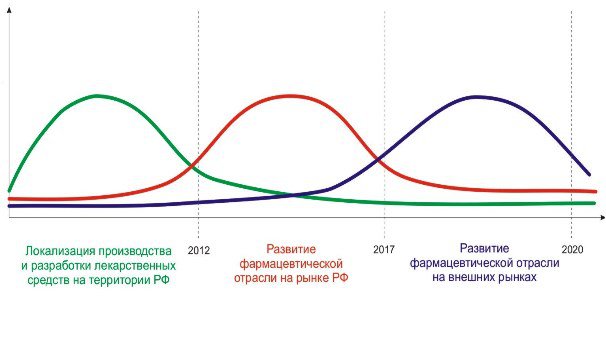
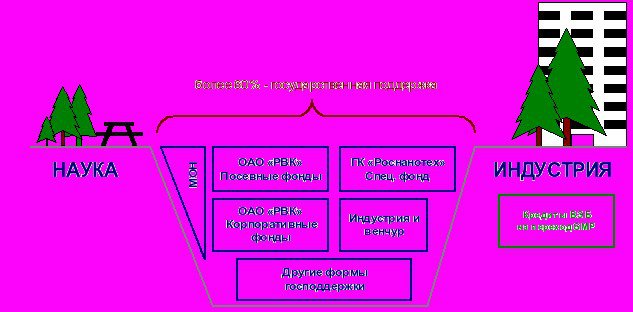
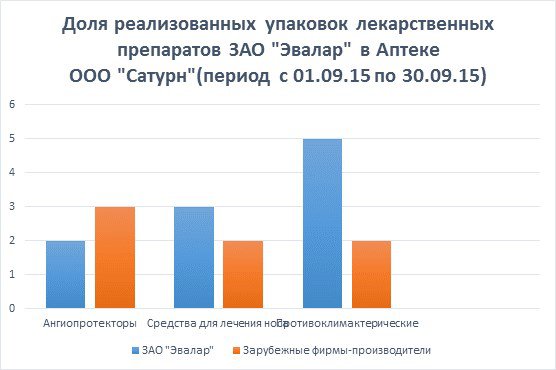
... стратегические цели, принципы и задачи развития фармацевтической промышленности, ситуацию в отрасли, проблемы отрасли, способы и пути решения указанных проблем.[12] 7.Исследование доли выпускаемой продукции отечественных производителей в ассортименте товаров аптек базы практики. 7.1.Анализ доли препаратов ЗАО «Эвалар» В Аптеке ООО «Сатурн», расположенной по адресу: Республика Марий Эл, пгт. ...
0 комментариев