Навигация
5. Добавление
Эти алгоритмы работают путем добавления элементов выборки в уже существующие кластеры. Ограниченность этих алгоритмов очевидна.
6. Поиск
Алгоритмы поиска обычно применяются к тем системам, для которых в результате математического анализа исключены многие из возможных способов разбиения на кластеры. С помощью этих алгоритмов производится такая оптимальная кластеризация системы, которая приводит к минимуму функции ошибок.
Существует много различных алгоритмов, однако ни один из них не приспособлен для решения любой из возникающих задач. Некоторые алгоритмы, например алгоритм ISODATA Болла и Холла [19,20] может осуществлять процедуры добавления, поиска, объединения и разбиения. Такие алгоритмы имеют более широкую область применения, однако ни один из них не является универсальным. К тому же многие алгоритмы являются эвристическими по своей природе, и поэтому успех их реализации, в конечном счете, зависит от мастерства исследователя. И наконец, последний недостаток методов кластеризации заключается в том, что иногда возникают трудности с отнесением неизвестного объекта к одному из уже имеющихся классов.
Несмотря на недостатки, методы кластеризации могут оказаться полезными для упорядочения систем, которые на первый взгляд кажутся совершенно неупорядоченными. Отметим также, что методы кластеризации необязательно требуют предварительной группировки объектов исследуемой выборки на классы. Алгоритмы кластеризации могут использоваться для выделения классов в выборках, способ классификации которых неочевиден. Как показано выше, алгоритмы кластеризации, основанные на различных способах определения расстояния, могут использоваться для расчета критериев подобия, для выделения существенных признаков и для преобразования исходных данных к виду, более удобному для дискриминантного анализа.
1.2.4 Программа PASS C&T
Знание известных биологически активных соединений и аналитические возможности даже самого лучшего из химиков - ограничены, и поэтому помощь специальной компьютерной системы в получении оценок по возможным видам биологической активности для различных классов соединений была бы полезной. Идея создания компьютерной системы прогноза биологической активности, на первый взгляд, выглядит достаточно просто: нужно собрать всю известную информацию о биологически активных соединениях, создать на этой основе обучающую выборку, провести анализ связей "структура-активность" для веществ из обучающей выборки и построить соответствующие зависимости. "Подставив" в эти зависимости данные о структуре нового вещества, можно получить в результате оценку его биологической активности.
Правда, традиционные подходы к анализу количественных соотношений "структура-активность" (КССА) применимы к соединениям одного и того же химического класса и, как правило, оперируют с одним видом биологической активности. Можно ли разработать подобные методы для веществ, гетерогенных как по химической структуре, так и по проявляемому ими биологическому действию?
Предложение предсказывать подобным образом спектр биологической активности вещества было впервые высказано в начале 70-х годов к.х.н. В.В. Авидоном c сотрудниками, работавшими тогда в НИИ по биологическим испытаниям химических соединений. В.В. Авидоном, совместно с к.х.н. В.Г. Блиновой, к.м.н. Е.М. Михайловским, Р.К. Казарян, к.ф.-м.н. В.С. Ароловичем и др., были разработаны оригинальные языки описания химической структуры, Тезаурус (структурированный словник) по биологической активности химических соединений, математические методы установления зависимостей "структура-активность" и прогноза свойств новых веществ; создан банк данных по биологически активным соединениям (обучающая выборка). На этой основе были осуществлены первые эксперименты по прогнозированию спектра биологической активности по структурной формуле вещества.
За истекшее двадцатилетие методы, первоначально предложенные для прогноза спектра биологической активности, претерпели существенные изменения. Эти изменения базируются как на теоретическом анализе методики прогнозирования, так и на имеющемся опыте ее применения для поиска веществ с требуемыми свойствами.
Современная версия компьютерной системы предсказания спектра биологической активности PASS C&T (Prediction of Activity Spectra for Substances: Complex & Training) реализована в 1998 году. Она включает в себя обучающую выборку, содержащую более 30000 биологически активных веществ с известной биологической активностью, и охватывает более 400 фармакологических эффектов, механизмов действия, а также мутагенность, канцерогенность, тератогенность и эмбриотоксичность.
Математический подход, используемый в PASS C&T, выбран Д.А. Филимоновым в результате сравнительного анализа 300 различных методов. Показано, что средняя точность прогноза с помощью PASS C&T при скользящем контроле с поочередным исключением по одному соединению из обучающей выборки составляет около 84%.
Результаты прогноза выдаются либо в виде текстового файла, который может в дальнейшем обрабатываться с помощью различных текстовых процессоров, либо в виде SDF файла, который может импортироваться в ISIS/Base и добавляться к имеющейся в базе данных информации о веществах. Далее обработка результатов прогноза осуществляется стандартными программными средствами, имеющимися в ISIS/Base.
Биологическая активность описывается в PASS C&T качественным образом ("да"/"нет"). Выдаваемые результаты прогноза помимо названий активности включают в себя оценки вероятностей наличия (Pa) и отсутствия каждой активности (Pi), имеющие значения от 0 до 1. Поскольку эти вероятности рассчитываются независимо, их сумма не равна единице.
Пример предсказания спектра биологической активности для препарата талидомид приведен ниже. Как видно из рисунка, известные для данного вещества виды активности (анксиолитическая, седативная, снотворная, тератогенная, модулятор цитокинов, ингибитор ангиогенеза, антагонист фактора некроза опухоли) содержатся в прогнозируемом спектре активности. Помимо этого, прогнозируется также ряд дополнительных видов активности – сердечно - сосудистый аналептик, антагонист нейрокинина, ингибитор кальпаина, и другие - которые указывают перспективные направления дальнейшего тестирования данного препарата.
Необходимо подчеркнуть, что для эффективного использования данные компьютерного прогноза должны рассматриваться специалистами с учетом имеющейся дополнительной информации.
Так, если целью исследования является поиск базовых структур лекарств, обладающих существенной, целесообразно отбирать из массива доступных веществ не те структуры, для которых величина Pa близка к единице (они могут оказаться близкими аналогами известных препаратов), а соединения с Pa<0.7. Риск получения отрицательного результата в эксперименте тем больше, чем меньше величина Pa, однако и новизна такой структуры (при подтверждении прогноза в эксперименте) будет более высокой.
Наоборот, если поставлена цель поиска близкого аналога известного препарата, то из массива имеющихся образцов следует отобрать вещества с наибольшими значениями Pa.
Кроме того, если, наряду с основным действием, известен перечень нежелательных побочных эффектов, то при отборе перспективных для исследований соединений можно руководствоваться комбинированным критерием:
- наличие в прогнозируемом спектре требуемых эффектов/механизмов;
- отсутствие нежелательных эффектов/механизмов.
Естественно, что при рассмотрении всего списка, включающего свыше 400 прогнозируемых видов активности, можно составить большое количество комбинаций из требуемых и нежелательных эффектов. Для их анализа сотрудник Лаборатории структурно-функционального конструирования лекарств НИИ Биомедхимии РАМН А.А. Лагунин разработал специальную компьютерную систему интерпретации спектров биологической активности веществ IBIAC, основанную на знаниях об известных взаимосвязях между фармакологическими эффектами и механизмом действия биологически активных веществ (более 2000 терминов, описывающих биологическую активность). С использованием системы IBIAC генерация перечня эффектов, соответствующих определенному механизму действия и, наоборот, списка вероятных механизмов, ответственных за проявление определенного эффекта, осуществляется автоматически.
Поскольку прогноз спектра биологической активности осуществляется на основе структурной формулы химического соединения, он может быть выполнен уже на этапе планирования синтеза. В итоге будут синтезированы лишь некоторые из теоретически возможных производных, в наибольшей степени удовлетворяющие критериям задачи.
Необходимо отметить, что прогноз спектра биологической активности возможен для низкомолекулярных органических соединений, структура которых не отличается принципиально от веществ обучающей выборки. Не имеет смысла прогноз для синтетических и биополимеров, для неорганических веществ и т.п.
Другое ограничение определяется необходимостью наличия не менее 5 веществ с известной активностью для формирования обучающей выборки. Очевидно, что в случае принципиально новых мишеней действия лекарственных препаратов, для которых имеются данные только об 1-2 лигандах, предсказание биологической активности таким методом не может быть реализовано.
Химическая структура и часть прогнозируемого спектра биологической активности для препарата талидомид (жирным шрифтом выделены активности, известные из эксперимента).
PASS CT 1.11 - Prediction of Activity Spectra for Substances
Copyright (c) 1998 V.V.Poroikov, D.A.Filimonov & Associates
Chemical Structure File: thalido.mol, <ACTIVITY_PREDICTION>
24 Substructure descriptors; 0 new, 84 Possible activities.
Pa Pi Activity
0.781 0.006 Cytokine modulator
0.713 0.019 Sedative
0.678 0.030 Cardiovascular analeptic
0.656 0.015 Angiogenesis inhibitor
0.439 0.007 Neurokinin antagonist
0.435 0.008 Calpain inhibitor
0.433 0.009 Oxytocin antagonist
0.443 0.024 Chemoprotective
0.421 0.011 Tumour necrosis factor antagonist
0.398 0.007 Hypnotic
0.439 0.050 NMDA agonist
0.407 0.028 Bronchodilator
0.430 0.059 Psychotropic
0.417 0.054 Anxiolytic
0.370 0.007 Protein kinase C inhibitor
0.428 0.068 Anticonvulsant
0.421 0.062 Teratogen
0.361 0.008 Antidiabetic symptomatic
0.377 0.035 Cardioprotectant
0.336 0.012 Benzodiazepine agonist partial
0.362 0.052 Spasmolytic, urinary
0.364 0.060 Analeptic
0.360 0.060 Nootropic
0.305 0.008 Uterine Relaxant
0.375 0.086 Septic shock treatment
0.385 0.102 Platelet adhesion inhibitor
В случае существенной по отношению к соединениям обучающей выборки новизны химической структуры прогнозируемого вещества (более 3-х дескрипторов ни разу не встретились в обучающей выборке) результаты прогноза могут иметь значительную погрешность. В этом случае целесообразно протестировать вещество на требуемые виды активности независимо от результатов прогноза, так как результатом может оказаться принципиально новая базовая структура.
В некоторых случаях вещество прогнозируется одновременно как агонист и антагонист (стимулятор и блокатор, активатор и ингибитор) по отношению к одним и тем же рецепторам (ферментам и т.п.). Это означает, что система не может дифференцировать внутреннюю активность вещества, а лишь указывает на его способность к связыванию с данным рецептором (ферментом).
И, наконец, необходимо иметь в виду, что система PASS C&T не может предсказать, станет ли конкретное вещество лекарственным препаратом, поскольку это будет зависеть также от многих других факторов (сравнительной оценки безопасности и клинической эффективности; наличия необходимых для разработки и внедрения инвестиций, и т.д.). Прогноз, однако, может помочь определить, какие тесты наиболее адекватны для изучения биологической активности конкретного химического вещества, и какие вещества из имеющихся в распоряжении исследователя наиболее вероятно проявят требуемые эффекты. [19]
1.3 Вывод
В этом и предыдущем разделах было дано краткое описание использовавшихся и используемых квантовохимических методов. Более детальное знакомство с ними практически не требуется для решения практических задач. Это связано с тем, что на основе анализа приближений, сделанных при разработке того или иного квановохимического метода, как правило, не удается установить область его применения и очертить круг задач, которые можно решить с его помощью. К сожалению, многие квантовохимические методы, которые лучше обоснованы с теоретической точки зрения, на практике дают плохие результаты и поэтому не применяются, а более грубые модели с удачно подобранными параметрами широко используются. Это связано с тем, что в любом квантовохимическом методе сделано достаточно много различных приближений. В некоторых методах ошибки, к которым приводят эти приближения, частично компенсируют друг друга и в результате получается хорошее согласие с экспериментом. Сказать заранее, будет или не будет иметь место такая компенсация нельзя, поэтому выяснить область применения и охарактеризовать точность конкретного метода можно лишь на основе численного эксперимента и систематизации опубликованного расчетного материала.
Глава 2. ВЫЧИСЛЕНИЕ ГЕОМЕТРИИ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ 2.1 Квантовохимические методы расчета
Данные статистического анализа результатов квантовохимических расчетов геометрических параметров стабильных органических молекул небольшого размера, содержащих одинарные и кратные связи, приведены в таблице 1.1 приложения А, показано, с какой точностью можно рассчитать геометрию молекул неэмпирическим методом и как меняются результаты расчетов в зависимости от выбора базиса. Обращает на себя внимание хорошее согласие с экспериментом, которое получается при использовании минимального и валентно - расщепленных базисов.
Сложнее обстоит дело с расчетом валентных углов. Если у молекулы нет неподеленных электронных пар, то расчет в валентно - расщепленном базисе приводит к хорошему согласию с экспериментом, но для расчета валентных углов в молекулах с неподеленными электронными парами в базис необходимо включить поляризационные орбитали.
Наряду с неэмпирическими методами для вычисления геометрии органических молекул широко используются и полуэмпирические методы. Среди них наиболее точные результаты для большинства типов соединений дают методы АМ1, МПДП и МЧПДП/3. Методом МПДП получается хорошее согласие с экспериментом практически для всех геометрических параметров молекул (см. табл. 1.2 приложение А).
2.1.1 Расчет потенциалов ионизацииПотенциалы ионизации органических молекул обычно вычисляют по теореме Купманса, которая связывает ПИ электрона с энергией хартри – фоковской орбитали исходной молекулы с замкнутой оболочкой. Для большинства соединений расчеты в этом приближении дают удовлетворительное согласие с экспериментальными вертикальными ПИ и поэтому широко используются для интерпретации данных фотоэлектронной спектроскопии. Кроме того, расчеты ПИ оп теореме Купманса используются для изучения реакционной способности некоторых органических соединений.
Наибольшее количество опубликованных расчетов ПИ выполнено методами МЧПДП/3 и МПДП. В таблице 1.3 приложения А приведены результаты расчетов ПИ методом МПДП.
Основное правило при расчете ПИ: если верхняя занятая молекулярная орбиталь у молекулы вырождена или почти вырождена, то механические деформации, которые снижают симметрию молекулы и снимают вырождение, приводят к уменьшению ее ПИ.
2.1.2 Расчет индексов реакционной способностиЭнергию межмолекулярного взаимодействия при сближении реагентов можно условно разбить на вклады трех типов: кулоновские, орбитальные и стерические. Энергия кулоновского взаимодействия зависит от распределения электронной плотности или от зарядов на атомах реагентов. Поэтому для некоторых реакций удается найти корреляцию между зарядами на атомах и выходом конечных продуктов реакции. Так, нуклеофильные реагенты присоединяются преимущественно к атомам, на которых локализованы большие положительные заряды, а электрофильные наоборот, - к атомам, на которых локализованы большие отрицательные заряды.
Корреляции между выходом конечных продуктов реакции и зарядами на атомах широко используются для объяснения экспериментальных данных. Обычно при вычислении заряда на атоме в квантовой химии пользуются анализом электронных заселенностей, предложенным Малликеном. В этом приближении заряд на атоме вычисляется по следующей формуле:
![]()
Здесь сумма берется по всем орбиталям i и j атома А; ZA – заряд ядра; Pij – матрица зарядов и порядков связей; Sij – матрица интегралов перекрывания. В полуэмпирических методах обычно пользуются упрощенной формулой:
![]()
Величины зарядов на атомах, вычисленные в этом приближении, в неэмпирических расчетах очень сильно зависят от выбора базиса, а в полуэмпирических – от выбора метода. Заряды на атомах, вычисленные в разных базисах и разными методами, могут различаться в 1,5 – 2 раза, но качественные результаты (знак и относительная величина заряда) обычно остаются одинаковыми. В неэмпирических расчетах заряды на атомах при расширении базиса обычно увеличиваются по абсолютной величине.
2.1.3 Вычисление теплот образованияТеплоты образования молекул являются фундаментальными термохимическими величинами. Однако их значение для многих органических соединений неизвестны, поэтому квантовохимические расчеты этих величин представляют большой интерес с точки зрения органической химии.
Параметры полуэмпирических методов МЧПДП/3 и МПДП подобраны так, чтобы наилучшим образом воспроизвести экспериментальные теплоты образования органических соединений при нормальных условиях. Средняя ошибка при вычислении теплот образования молекул методом МЧПДП/3 составляет 38 кДж/моль, а методом МПДП – 25 кДж/моль.
Сложнее вычислить теплоты образования и теплоты атомизации молекул неэмпирическими методами. Даже для небольших молекул неэмпирический расчет в базисе 6-31ГФ* приводит к ошибкам в теплотах образования, превышающим 100 кДж/моль. Это связано с неполнотой использованного базиса и неучетом энергии электронной корреляции. С увеличением размера молекулы ошибки в неэмпирически вычисленных теплотах образования хотя и возрастают, но в значительной мере носят систематический характер. Поэтому их можно уменьшить с помощью коррекции конечных результатов по аддитивной схеме.
Расчеты с эмпирически подобранными значениями параметров показали, что с их помощью можно уменьшить ошибку при вычислении теплот образования органических молекул: при использовании базиса ОСТ-3ГФ – до 45 кДж/ моль, а при использовании базисов 3-21ГФ и 6-31ГФ* - соответственно до 29 и 25 кДж/моль (табл. 1.4 приложения А).
Такие поправки нельзя использовать при расчете поверхностей потенциальной энергии, так как в ходе реакции всегда образуются структуры, в которых одни связи частично разорваны, а другие частично образованы, и нельзя сказать, между какими атомами есть валентная связь, а между какими ее нет. Однако поправки такого типа можно использовать для расчета тепловых эффектов реакций и для решения многих других прикладных задач.
2.1.4 Расчет тепловых эффектов органических реакцийВеличина теплового эффекта позволяет оценить термодинамическую возможность протекания химической реакции или отдельной элементарной стадии. В общем случае теплота реакции не коррелирует с ее скоростью. Поэтому данные о тепловых эффектах широко применяются для изучения реакционной способности органических соединений. Следует, однако, отметить, что расчет тепловых эффектов для квантовой химии является весьма сложной задачей, так как эту величину необходимо знать с точностью до 4 кДж/моль (химическая точность).
Из полуэмпирических методов наиболее широко используются схему МЧПДП/3 и МПДП, причем метод МПДП дает более точные результаты. В таблице 1.6 приложения А приведены результаты расчетов этим методом тепловых эффектов некоторых реакций изомеризации. Из этих данных видно, что для некоторых реакций согласие теории с экспериментом хорошее, но в отдельных случаях ошибка получается очень большой.
Результаты неэмпирических расчетов тепловых эффектов органических реакций очень сильно зависят от выбора метода. В валентно – расщепленных базисах для реакций с участием насыщенных молекул ошибки составляют около 40 кДж/моль, но, если в молекуле есть кратные связи или молекулы являются напряженными, ошибки обычно увеличиваются приблизительно в 2 раза.
2.1.5 Расчет поверхностей потенциальной энергииДля получения наиболее полной информации о механизме реакции необходимо вычислить многомерную поверхность потенциальной энергии (ППЭ), то есть рассчитать зависимость полной энергии от координат атомных ядер. Наиболее интересными и важными при изучении механизма реакции являются так называемые стационарные точки на ППЭ. Под этим термином понимают минимумы и седловые точки на ППЭ (рисунок 3.1.5.1, 3.1.5.2) В стационарных точках производные полной энергии по всем независимым координатам равны нулю.


Рисунок 2.1.5.1 Стационарные точки
а — минимум локальный или глобальный;
б — седловая точка

Рисунок 2.1.5.2 Простейшая ППЭ
Темные кружки – исходные
реагенты и продукты реакции;
крестик — переходное состояние.
В точке минимума полной энергии матрица вторых производных имеет только положительные собственные значения, а в седловой точке – одно отрицательное собственное значение. Минимумы полной энергии соответствуют устойчивым структурам и интермедиатам, а седловые точки – переходным состояниям.
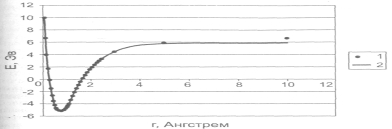
Рисунок 2.1.5.3. Зависимость потенциальной энергии молекулы водорода от расстояния между атомами: 1 — расчет полуэмпирическим методом РМЗ; 2 — аппроксимация потенциалом Морзе.
Типичный вид простейшей двумерной ППЭ показан на рисунке 2.1.5.2. Здесь минимумы соответствуют исходным реагентам и конечным продуктам реакции, а седловая точка – переходному состоянию. Минимумы на рисунке соединены пунктирной линией, которая проходит по дну долины на ППЭ через седловую точку. Эта линия показывает путь реакции в двумерном пространстве или траекторию движения реагентов в ходе реакции. Для большинства реакций ППЭ имеют более сложный вид.
В таблицах 1.7 и 1.8 приложения А сопоставлены данные расчета параметров переходных состояний для реакций, изображенных на схемах 1 - 5, методами МПДП и КМПДП (метод МПДП с учетом электронной корреляции), неэмпирическим методом в приближении Хартри – Фока без учета и с учетом электронной корреляции.
Схема I Схема II Схемa III
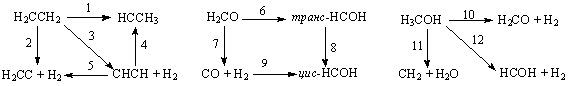
Схема IV Схемa V
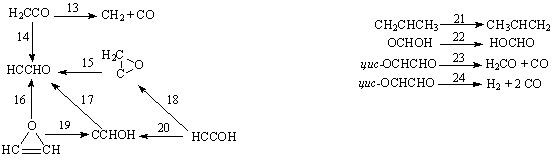
В таблицах неэмпирический расчет в приближении Хартри – Фока без учета электронной корреляции обозначен ХФ, с учетом электронной корреляции – КХФ. Для реакций, изображенных на схемах 1 – 3, расчеты с оптимизацией геометрии в приближении Хартри – Фока проведены в базисе 6-31ГФ*, для реакций, изображенных на схемах 4, 5, - в базисе 3-21ГФ или 4-31ГФ. Электронная корреляция учитывалась только при вычислении энергии активации.
Из этих данных видно, что геометрические параметры переходных состояний, вычисленные методами МПДП и КМПДП, находятся в хорошем согласии с данными неэмпирических расчетов без учета электронной корреляции.
Вопрос о влиянии электронной корреляции на геометрию переходных состояний был рассмотрен в работе Шредера [20]. В ней методом МПДП без учета и с учетом электронной корреляции была рассчитана геометрия переходных состояний для реакций, изображенных на схемах 1 – 5, и показана хорошая сходимость с экспериментом.
2.1.6 Силовые постоянные химических связей и частоты внутримолекулярных колебанийДля расчета силовых постоянных довольно широко применяют как полуэмпирические, так и неэмпирические методы квантовой химии. В любом случае сначала оптимизируют геометрию, то есть определяют наиболее устойчивую конформацию, отвечающую минимуму полной энергии; затем вычисляют вторые производные полной энергии по естественным координатам, а при необходимости – кубичные и биквадратные члены.
При использовании минимального слейтеровского базиса согласие с экспериментом получается весьма посредственное. Для полуэмпирических методов характерно относительное занижение частот деформационных колебаний по сравнению с валентными. В связи с тем, что ошибки в большинстве случаев носят систематический характер, их удается значительно уменьшить введением эмпирически подобранных масштабных корректирующих множителей для определенных типов силовых постоянных или инкременентов, которые прибавляются к рассчитанным частотам.
Для расчетов методами МЧПДП/3 и МПДП Дьюар и Форд [23] подобрали систему инкрементов, специфичных для валентных, деформационных и торсионных колебаний определенных атомных групп или связей; на очень большом числе примеров продемонстрирована удовлетворительная точность результатов.
сульфаниламид квантовый химический органический молекула
Более логичным представляется корректирование значений силовых постоянных, и на этом пути достигнуты положительные результаты. В настоящее время используется несколько методик подбора корректирующих множителей. Наиболее распространенными являются следующие предложения:
0 комментариев