ОПРЕДЕЛЕНИЕ НЕМЕТАЛЛИЧЕСКИХ ЭЛЕМЕНТО6
Фотометрический фенол-гипохлоритный
Фотометрический метод с реактивом
НИТРАТЫ
Определение восстановлением амальгамой
НИТРИТЫ
Ч. д. а, в бидистилляте, разбавляют до 100 мл бидистиллятом и прибавляют 0,2-0,5 мл реактива Несслера. Раствор можно применять после осветления
Навигация
Ч. д. а, в бидистилляте, разбавляют до 100 мл бидистиллятом и прибавляют 0,2-0,5 мл реактива Несслера. Раствор можно применять после осветления
Анализ азота и его соединений
116208
знаков
7
таблиц
6
изображений
0 ч. д. а, в бидистилляте, разбавляют до 100 мл бидистиллятом и прибавляют 0,2-0,5 мл реактива Несслера. Раствор можно применять после осветления.
Комплексен III, 50%-ный раствор. Растворяют 10 г NaOH ч. д. а, в 60 мл бидистиллята и в полученном растворе растворяют 50 г комплексона III. Смесь доводят бидистиллятом до 100 мл.
Едкий натр, ч. д. а., 15%-ный раствор в бидистилляте. Хлорид аммония, стандартный раствор.
Раствор А. Растворяют 0,2965 г NH4Cl ч. д. а, в бидистилляте и разбавляют до 1 л. В 1 мл раствора содержится 0,100 мг NH+4. Раствор должен быть свежеприготовленным.
Раствор Б. Доводят 50,0 мл рабочего раствора А бидистиллятом до 1 л, в 1 мл раствора содержится 0,005 мг NH+4. Раствор должен быть свежеприготовленным. Реактивы не должны содержать аммиак, что контролируется холостым опытом.
Калибровочная кривая. В мерные колбы емкостью 50 мл или в цилиндры Несслера с меткой на 50 мл последовательно прибавляют 0; 0,5; 1,0; 2,0; 4,0; 6,0; 8,0; 10; .; 40 мл рабочего стандартного раствора Б и объем доводят бидистиллятом до 50 мл. Полученные растворы с концентрациями 0; 0,20; 0,40; 0,60; 0,80; 1,0; ..., 4,0 мг-ион NH+4 в 1 л обрабатывают описанным ниже способом и строят график зависимости оптической плотности от содержания аммиака; вводят поправку на холостой опыт. При сравнении в цилиндрах Несслера приготовляют шкалу только до концентрации NH+4 2 мг!л.
Ход определения. К 50 мл первоначальной пробы, или к 50 мл осветленной пробы, или к меньшему ее объему, доведенному до 50 мл бидибидистиллятом, прибавляют 1-2 капли раствора комплексона III или сегнетовой соли и смесь тщательно перемешивают. При анализе очень жестких вод нужно прибавить 0,5-1 мл раствора сегнетовой соли или комплексона III. Затем прибавляют 1 мл реактива Несслера и снова перемешивают. По истечении 10 мин колориметрируют или сравнивают с серией стандартов, приготовленных в цилиндрах Несслера. Окраска смеси не изменяется в течение 30 мин. Из величины оптической плотности вычитают оптическую плотность холостого опыта. Если нужно, вычитают и оптическую плотность пробы, к которой вместо реактива Несслера прибавляют 1 мл 15%-нога раствора едкого натра, и по калибровочному графику находят содержание аммиака.
Расчет. Содержание NH+4 мг/л(х) или в мг-экв/л (у) вычисляют по формулам:
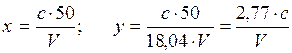
где с - найденная концентрация NH+4, мг1л; V - объем пробы, взятой для анализа, мл; 18,04 - эквивалент NH+4; 50 - объем пробы, мл.
Округление результатов
| Диапазон, мг/л | 0,05-2,00 | 2,0-5,0 | 5,0-10,0 | 10,0-20,0 |
| Округление ....... |
|
|
|
|
| в мг/л | 0,05 | 1,0 | 0,2 | 0,5 |
| в мг-экв/л | 0,002 | 0,005 | 0,01 | 0,02 |
Определение с перегонкой
Аммиак перегоняют из щелочной среды и определяют в дистилляте колориметрическим или объемным методом. Для выделения аммиака можно применять обыкновенную перегонку или перегонку с водяным паром, например в аппарате Парнаса - Вагнера. Пробу подщелачивают либо фосфатным буферным раствором до рН 7,4 (питьевые, поверхностные, биологически очищенные сточные воды и т. п.), либо раствором едкого натра (фенольные сточные воды, сточные воды коксохимических заводов и т. п.). Подщелачивание едким натрем неприменимо для вод, содержащих белковые или другие вещества, выделяющие аммиак.
В зависимости от содержания в пробе аммиака для его поглощения в приемнике применяют серную кислоту различной нормальности или борную кислоту. Количество серной кислоты, израсходованной на нейтрализацию перегнанного аммиака, определяют обратным титрованием раствором едкого натра. При использовании борной кислоты перегнанный аммиак определяют прямым титрованием раствором серной кислоты. Титруют по метиловому красному или со смешанным индикатором, состоящим из метилового красного и метиленовой синей.
Колориметрическое определение аммиака в дистилляте с реактивом Несслера возможно в том случае, если концентрация аммиака в 100 мл дистиллята меньше 0,1 мг. При более высокой концентрации аммиака применяется объемный метод. Титруют 0,02 н., 0,1 н. и 1 н. растворами кислот при содержании аммиака в 100 мл дистиллята, равном 5, 25 и более 25 мг.
Мешающие влияния. Определению мешает свободный хлор; его устраняют перед перегонкой прибавлением тиосульфата или арсенита натрия (см. «Колориметрическое определение с реактивом Несслера»).
Кальций в концентрациях, превышающих 250 мг/л, оказывает влияние на установление pH. В этом случае раствор подщелачивают 40 мл буферного фосфатного раствора и смесь обрабатывают кислотой или щелочью для получения рН 7,4.
Летучие органические соединения, которые мешают колориметрическому определению аммиака в дистилляте, устраняют кипячением слабо подкисленной пробы.
Аппаратура
Перегонный аппарат для обычной перегонки или аппарат Парнаса-Вагнера для перегонки с водяным паром.
Реактивы
Бидистиллят, не содержащий аммиака.
Буферный раствор, рН 7,4. Растворяют 14,3 а КН2РО4 (безводного) ч. д. а. и 68,8 г К2НРО4 (безводного) ч. д. а, в бидистилляте и разбавляют до 1 л. Значение рН 7,4 при необходимости корректируют добавлением едкого кали или соляной кислоты.
Едкий цитр, 40%-ный раствор. Растворяют 400 г NaOH ч. д. а, в бидистилляте и разбавляют до 1 л.
Серная кислота, приблизительно 1 н. раствор. Прибавляют к 500 мл бидистиллята 28 мл H2SO4 ч. д. а., концентрированной и после перемешивания разбавляют до. 1 л.
Серная кислота, приблизительно 0,1 н. раствор. Отмеривают 2,8 мл концентрированной H2SO4 ч. д. а, и добавляют к 500 мл бидистиллята, перемешивают и разбавляют до 1 л.
Серная кислота, 0,02 н. раствор. Разбавляют 200 мл 0,1 н. H2SO4 бидистиллятом и доводят до 1 л. При использовании этого раствора для титрования титр или поправку проверяют титрованием 0,02 н. раствором NaOH.
Едкий натр 1 н. раствор. Растворяют 40 г NaOH ч. д. а, в бидистилляте и разбавляют до 1 л. Титр или поправку определяют титрованием 1 н. раствором щавелевой кислоты.
Едкий натр, 0,1 н. раствор. Растворяют 4 г NaOH ч. д. а, в холодном свеже- прокипяченном бидистилляте и разбавляют до 1 л. Титр или поправку определяют титрованием раствором щавелевой или другой кислоты той же нормальности (см. стр. 63).
Едкий натр, 0,02 н. раствор. Разбавляют 200 мл 0,1 н. титрованного раствора едкого натра холодным свежепрокипяченным бидистиллятом до 1 л. Определяют титр или поправку так же, как для 0,1 н. раствора NaOH. Титруют 0,02 н. раствором щавелевой кислоты, который приготовляют разбавлением 200 мл 0,1 н. раствора щавелевой кислоты до 1 л свежеприготовленным и охлажденным бидистиллятом при 20°С.
Борная кислота, 2%-ный раствор. Растворяют 20 г H3BO3 ч. д. а, в бидистилляте и разбавляют до 1 л.
Метиловый красный, 0,1%-ный раствор. Растворяют 0,1 а натриевой соли метилового красного в 100 мл 96%-наго спирта.
Смешанный индикатор. Смешивают 2 объема 0,2%-наго раствора метилового красного в 96%-ном спирте с 1 объемом 0,2%-наго раствора метиленовой синей в 96%-ном спирте.
Реактив Несслера (приготовление - см. «Колориметрическое определление с реактивом Несслера»).
Ход определения. В перегонную колбу отмеривают 10-500 мл, в зависимости от ожидаемого содержания аммиака, пробы, которую при необходимости разбавляют бидистиллятом приблизительно до 250 мл. Если необходимо, смесь нейтрализуют 1 н. раствором едкого натра (определяется индикаторной бумагой). Пробы, которые подще- лачивались при отборе, нейтрализовать не надо.
В приемник, который представляет собой колбу для титрования емкостью 500 мл, наливают 25 мл серной кислоты соответствующей нормальности или 25 мл 2%-ного раствора борной кислоты и доливают бидистиллят так, чтобы конец холодильника был погружен в жидкость.
В перегонную колбу прибавляют 100 мл фосфатного буферного раствора. Значение рН смеси в колбе не должно отличаться более чем на ±0,2 от рН 7,4. Для установления рН в некоторых случаях приходится прибавлять большее количество буферного раствора - до 40 мл на 100 мл пробы (биологически очищаемые сточные воды
и др.). Фекальные и сточные воды не подщелачивают буферным раствором, а добавляют 20 мл 40%-ного раствора едкого натра.
В приемник отгоняют около 200-100 мл жидкости. При использовании аппарата Парнаса - Вагнера в приемник перегоняют с водяным паром не менее 50 мл дистиллята, опускают приемник и собирают по каплям еще около 5 мл дистиллята. Затем прерывают перегонку и конец, холодильника ополаскивают бидистиллятом.
Аммиак, перегнанный в приемник, определяют объемным или колориметрическим методом. При определении объемным методом с применением серной кислоты дистиллят титруют раствором едкого натра соответствующей концентрации по метиловому красному или со смешанным индикатором. Титровать надо раствором той же нормальности, какой был применен для поглощения аммиака. Параллельно определяют количество раствора щелочи, расходуемое на титрование 25 мл кислоты, помещенной в приемник. Титрование ведут до получения такой же окраски индикатора.
При определении объемным методом с применением борной кислоты проводят холостой опыт (25 мл раствора борной кислоты титруют 0,02 н. раствором серной кислоты по метиловому красному или со смешанным индикатором). Дистиллят титруют тем же титрованным раствором до такой же окраски, какая была получена при титровании холостого опыта.
При колориметрическом определении содержимое приемника обрабатывают 1 н. кислотой до рН 6, переливают в мерную колбу емкостью 250 мл и объем доводят до метки бидистиллятом. В 50 мл раствора определяют аммиак способом, указанным при непосредственном колориметрическом определении аммиака реактивом Нес- слера .
Расчет. Содержание NH+4 в мг/л (х) или мг-экв (у) вычисляют следующими способами.
При определении объемным методом с поглощением серной кислотой по формулам:
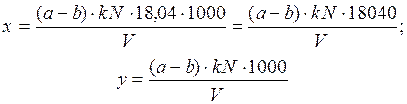
где а - объем раствора едкого натра, израсходованного на титрование 25 мл раствора серной кислоты, мл; b - объем раствора едкого натра, израсходованного на титрование дистиллята, мл; k - поправочный коэффициент к нормальности титрованного раствора едкого натра; N - нормальность титрованного раствора едкого натра; V - объем пробы, взятой для определения, мл; 18,04 - эквивалент NH+4.
При определении объемным методом с поглощением борной кислотой вычисление проводят по тем же формулам, используя следующие обозначения: а - объем 0,02 н. раствора серной кислоты, израсходованного на титрование дистиллята, мл; b - объем 0,02 н. раствора серной кислоты, израсходованного на титрование в холостом опыте, мл; k - поправочный коэффициент к нормальности титрованного раствора серной кислоты; N - нормальность титрованного раствора серной кислоты .
При определении колориметрическим методом вычисляют по фор мулам:
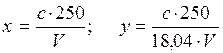
где с - концентрация аммиака, найденная во калибровочному графику, мгал. V - объем пробы, взятой для определения при перегонке, мл; 18,04 - эквивалент NH+4; 250 - объем разбавленной пробы.
Округление результатов. Результаты, полученные при объемном определении, округляют до 0,1 мг в области до 10 мг/л и до целых миллиграммов в области до 100 мг/л и т. д. Результаты колориметрического определения округляют так же, как при непосредственном колориметрическом определении с реактивом Несслера.
По найденному общему содержанию аммиака можно рассчитать концентрации аммиака и NH+4, если известно значение рН воды. Найденное общее содержание аммиака надо выразить в мг-экв/л и по табл. 2 найти концентрации NH3 и NH+4 в мг-экв/л. Умножив полученные величины соответственно на 17,03 и 18,04, можно получить концентрации NH3 и NH+4 в мг/л.
Таблица 2. Относительное содержание NH3 в воде, %
|
| pH | ||||||||
| t,°C | 6 | 7 | 8 | 8,5 | 9 | 9,5 | 10 | 10,5 | 11 |
| 25 | 0,05 | 0,49 | 4,7 | 13,4 | 32,9 | 60,7 | 83,1 | 93,9 | 98,0 |
| 15 | 0,02 | 0,23 | 2,3 | 6,7 | 19,0 | 42,6 | 70,1 | 88,1 | 96,0 |
| 5 | 0,01 | 0,11 | 0,9 | 3,3 | 9,7 | 25,3 | 51,7 | 77,0 | 91,5 |
Содержание NH+4 (в %) находят, вычитая из 100% указанное в таблице содержание NH3.
Таблица составлена для ионной силы раствора, равной 0,025, что приблизительно отвечает общему содержанию солей, равному 1 г/л. Колебания в величине ионной силы сравнительно мало отражаются на представленных в таблице величинах. Влияние температуры весьма значительно, особенно при средних значениях pH.
НИТРИТЫ
Нитриты являются промежуточным продуктом биохимического окисления аммиака или восстановления нитратов. Их присутствие свидетельствует о фекальном загрязнении вод. В поверхностных водах нитриты быстро переходят в нитраты. Они присутствуют в концентрациях от нескольких микрограммов до десятых долей миллиграмма в 1 л. В большем количестве они находятся в некоторых промышленных и биологически очищенных сточных водах.
Для определения нитритов в питьевых, поверхностных н сточных водах предлагается колориметрический метод с сульфаниловой кислотой и ![]() -нафтиламином.
-нафтиламином.
Вследствие нестойкости нитритов их надо определять сразу же после отбора пробы. Если это невозможно, пробу консервируют добавлением 1 мл концентрированной H2SO4, или 2-4 мл хлороформа на 1 л. Можно также охлаждать пробу до 3-4° С.
Результаты определения выражают в миллиграммах нитрит- ионов в 1 л, а при больших концентрациях - в мг-экв в 1 л: 1 мг ![]() = 0,02174 мг-экв
= 0,02174 мг-экв ![]() ; 1 мг-экв
; 1 мг-экв ![]() = 46,005 мг
= 46,005 мг ![]() .
.
Качественное определение
К 10 мл пробы прибавляют 1 мл раствора сульфаниловой кислоты и 1 мл раствора а-нафтиламина (приготовление - см. ниже). В присутствии нитритов появляется розовая или красно-фиолетовая окраска. Чувствительность определения составляет около 0,01 мг нитритов в 1 л воды.
Колориметрическое определение с сульфаниловой кислотой
и ![]() -нафтиламином
-нафтиламином
Метод основан на диазотировании сульфаниловой кислоты присутствующими в пробе нитритами и реакции полученной соли с ![]() -нафтиламином с образованием красно-фиолетового азокрасителя. Интенсивность окраски пропорциональна концентрации нитритов. Протекание реакции в значительной степени зависит от рН среды.
-нафтиламином с образованием красно-фиолетового азокрасителя. Интенсивность окраски пропорциональна концентрации нитритов. Протекание реакции в значительной степени зависит от рН среды.
Без разбавления пробы можно определять визуально от 0,002 до 0,025 мг ![]() в 1 л, колориметрически, в зависимости от применяемого фотометра, - от 0,001 до 0,6 мг1л. Точность определения ±0,002 мг/л.
в 1 л, колориметрически, в зависимости от применяемого фотометра, - от 0,001 до 0,6 мг1л. Точность определения ±0,002 мг/л.
Мешающие влияния. Определению мешают взвешенные вещества и мутность воды. Поэтому перед анализом пробу необходимо профильтровать. Если мутность фильтрованием не устраняется и сточные или сильно загрязненные воды содержат коллоидные вещества, необходимо пробу осветлить коагулированном гидроокисью алюминия. Для этого к 100 мл пробы прибавляют около 0,5 г активированного угля, 1 мл 12,5%-нога раствора сульфата алюминия и калия КАl(SO4)2•12Н2O и раствор аммиака до получения рН 5,8. После взбалтывания дают осадку осесть до полного осветления пробы. Фильтруют через сухой плотный фильтр (синяя лента). Для определения берут часть фильтрата. Осветление можно проводить также взбалтыванием 100 мл пробы с 2 мл суспензии гидроокиси алюминия (приготовление суспензии - см. «Колориметрическое определение с фенолдисульфоновой кислотой»).
В анализируемой пробе не должны присутствовать сильные окислители или восстановители, которые мешают определению.
Железо (III), ртуть (II), серебро, висмут, сурьма (III), свинец, золото (III), хлороплатинаты и метаванадаты мешают определение), так как выпадают в осадок. Влияния их устраняют соответствующим разбавлением.
Медь (II) понижает результаты вследствие вызываемого ею каталитического распада диазотированной сульфаниловой кислоты. В присутствии меди пробу также разбавляют.
Определению мешает и окраска воды. Слабое окрашивание и незначительная мутность питьевых и поверхностных вод при определении оптической плотности компенсируются холостым раствором, в котором к пробе прибавляют только раствор сульфаниловой кислоты.
Определению мешает и трихлорамин. При добавлении реактивов в обратном порядке его мешающее влияние можно в некоторой степени снизить.
Аппаратура
Фотометр с зеленым светофильтром (![]() = 520 нм).
= 520 нм).
Кюветы с толщиной слоя 1-5 см или набор Цилиндров Несслера емкостью 50 мл.
Реактивы
Сульфаниловая кислота, 0,6%-ный раствор. Растворяют 6,0 г сульфаниловой кислоты ч. д. а, в 750 мл горячей дистиллированной воды. К полученному раствору прибавляют 250 мл ледяной уксусной кислоты.
![]() -Нафтиламин, 0,6%-ный раствор. Растворяют 1,2 г нафтиламина ч.д.а. в дистиллированной воде, прибавляют 50 мл ледяной уксусной кислоты и доводят дистиллированной водой до 200 мл. При образовании мути раствор фильтруют через хлопчатобумажную ткань, промытую дистиллированной водой. Раствор сохраняется 2-3 месяца.
-Нафтиламин, 0,6%-ный раствор. Растворяют 1,2 г нафтиламина ч.д.а. в дистиллированной воде, прибавляют 50 мл ледяной уксусной кислоты и доводят дистиллированной водой до 200 мл. При образовании мути раствор фильтруют через хлопчатобумажную ткань, промытую дистиллированной водой. Раствор сохраняется 2-3 месяца.
Нитрит натрия, стандартный раствор. Растворяют 0,1497 г NaNO2 ч.д. а., высушенного при 105° С, в дистиллированной воде (лучше в стерилизованной) и доводят водой до 1 л. Раствор консервируют добавлением 1 мл хлороформа и сохраняют в холодном месте; он устойчив в течение месяца. В 1 мл этого раствора содержится 0,100 мг ![]() .
.
Рабочий раствор I. Разбавляют 100 мл основного раствора дистиллированной водой до 1 л. Раствор должен быть всегда свежеприготовленным. В 1 мл этого раствора содержится 0,010 мг ![]() .
.
Рабочий раствор II. Разбавляют 50 мл рабочего раствора 1 дистиллированной водой до 1 л. Раствор должен быть всегда свежеприготовленным. В 1 мл раствора содержится 0,0005 мг ![]() .
.
Калибровочная кривая. Для построения калибровочной кривой берут серию из шести или восьми стандартных растворов (соответственно применяемому прибору) с концентрацией нитрит-ионов в пределах от 0 до 0,60 мг/л. Строят график зависимости оптической плотности от концентрации нитрит-ионов.
Шкала стандартов. При визуальном определении одновременно с пробой приготовляют серию стандартных растворов в цилиндрах Несслера. В цилиндры емкостью 50 мл отмеривают пипеткой 0; 0,20; 0,50; 1,0; 1,5; 2,0 и 2,5 мл стандартного Рабочего раствора 11 и объемы доводят дистиллированной водой до 50 мл. Далее поступают так же, как при анализе пробы. Серия стандартных растворов соответствует концентрациям 0,000; 0,002; 0,005; 0,010; 0,015; 0,020 и 0,025 мг ![]() /л.
/л.
Ход определения. Для определения берут 50 мл или меньшее количество профильтрованной пробы и доводят до 50 мл дистиллированной водой. Прибавляют 1 мл раствора сульфаниловой кислоты
и смесь тщательно перемешивают. Если проба мутная или окрашенная, определяют ее оптическую плотность и затем вычитают из оптической плотности пробы. После пятиминутного стояния прибавляют 1 мл раствора ![]() -нафтиламина и смесь снова перемешивают. Пробу колориметрируют или сравнивают с серией стандартов, приготовленных в цилиндрах Несслера, через 40 мин после прибавления раствора
-нафтиламина и смесь снова перемешивают. Пробу колориметрируют или сравнивают с серией стандартов, приготовленных в цилиндрах Несслера, через 40 мин после прибавления раствора ![]() -нафтиламина и по калибровочной кривой находят содержание нитритов.
-нафтиламина и по калибровочной кривой находят содержание нитритов.
Расчет. Содержание нитрит-ионов (х) в мг/л или (у) в мг-экв/л вычисляют по формулам:
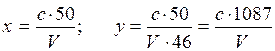
где с - концентрация нитрит-ионов, найденная по калибровочному графику или по шкале стандартов, мг/л; V - объем пробы, взятой для определения, мл; 50 - объем, до которого разбавлена проба мл, 46 - эквивалент нитрит-иона ![]() .
.
НИТРАТЫ
Нитраты встречаются почти во всех видах вод. В поверхностных и родниковых водах количество их обычно незначительно. Однако в некоторых родниковых водах концентрация нитратов высока. .
Большое количество нитратов указывает иногда на загрязнение в прошлом фекальными водами. Определение нитратов в грунтовых водах служит оценкой характера процессов минерализации при фильтровании воды через почвенные слои. При исследовании поверхностных вод по содержанию нитратов можно судить о протекающих процессах самоочистки, а при биологической очистке сточных вод - о процессе нитрификации. Некоторые промышленные сточные воды содержат значительные количества нитратов.
Методика определения нитратов еще окончательно не разработана. На основании опыта можно сказать, что для анализа питьевых, поверхностных и очищенных сточных вод, содержащих 0,5-50 мг1л нитратов, наиболее пригоден колориметрический метод с фенол- дисульфоновой кислотой. Хорошие результаты дает и колориметрический метод с салицилатом натрия. Этим методом определяются 0,1-20 мг/л нитратов.
Для рядовых серийных анализов проб, содержащих 5-30 мг1л нитратов, можно применять полярографический метод.
Большие количества нитратов можно определять указанными методами при соответствующем разбавлении пробы.
Метод восстановления нитратов сплавом Деварда до аммиака с последующей перегонкой может быть использован при анализе
сточных вод, содержащих нитраты в количествах, превышающих 5 мг/л.
Колориметрический метод с бруцином применим для определения 1-20 мл ![]() в 1 л.
в 1 л.
Если проба не была обработана в день отбора, ее хранят в холодильнике или консервируют добавлением 1 мл концентрированной серной кислоты или 2-4 мл хлороформа на 1 л пробы.
Результаты определений выражают в мг-екв/л или в мг/л ![]() ; 1 мг-экв
; 1 мг-экв ![]() = 62,00 мг
= 62,00 мг ![]() , 1 мг
, 1 мг ![]() = 0,01613 мг-экв
= 0,01613 мг-экв ![]() .
.
Качественное определение.
К 5 мл концентрированной серной кислоты в пробирке при постоянном перемешивании прибавляют по каплям 2 мл испытуемой воды. После этого вводят незначительное количество твердого бруцина (Осторожно, сильный яд!) и смесь снова перемешивают. Появившееся желтое или коричнево-красное окрашивание указывает на присутствие нитратов. Чувствительность реакции 1 мг ![]() в 1 л и более. Реакцию можно использовать для ориентировочного определения необходимого последующего разбавления пробы. Применяемая серная кислота не должна содержать нитратов.
в 1 л и более. Реакцию можно использовать для ориентировочного определения необходимого последующего разбавления пробы. Применяемая серная кислота не должна содержать нитратов.
Колориметрическое определение
с фенолдисульфоновой кислотой.
Определение основано на образовании желтого соединения в результате реакции нитратов с фенолдисульфоновой кислотой. Без разбавления этим методом можно определять от 0,5 до 50 мг ![]() в 1 л воды.
в 1 л воды.
При концентрации ![]() 2-50 мг1л и отсутствии мешающих влияний точность определения ±0,5 мг!л.
2-50 мг1л и отсутствии мешающих влияний точность определения ±0,5 мг!л.
Мешающие влияния. Определению мешают хлориды. Концентрация их в пробе не должна превышать 10 мг/л. Уменьшить их концентрацию можно соответствующим разбавлением. Мешающее влияние хлоридов можно также устранить добавлением сульфата серебра (см. ниже). Однако при анализе некоторых вод избыток ионов серебра вызывает окрашивание раствора или образование мути. Для подщелачивания пробы применяют едкое кали или аммиак.
Если для осаждения хлоридов был применен сульфат серебра, то подщелачивать раствор едким кали нельзя, так как этот реактив вызывает коричневое окрашивание; вместо него следует применять раствор аммиака.
При содержании нитритов, превышающем 0,7 мг/л, получаются повышенные результаты. Окрашенные соединения не должны присутствовать.
Удаление хлоридов. Определяют в пробе содержание хлоридов. К 100 мл пробы прибавляют эквивалентное количество раствора сульфата серебра (4,40 г Ag2SO4 ч. д. а., свободного от нитритов, растворяют в дистиллированной воде и доводят объем до 1 л, 1 мл раствора соответствует 1 мг ![]() ). Осадок хлорида серебра отфильтровывают или отделяют центрифугированном после предварительного нагревания смеси. Целесообразно оставить смесь на ночь в темноте.
). Осадок хлорида серебра отфильтровывают или отделяют центрифугированном после предварительного нагревания смеси. Целесообразно оставить смесь на ночь в темноте.
Удаление окраски. К 150 мл пробы прибавляют 3 мл суспензии гидроокиси алюминия (125 г сульфата алюминия и калия или сульфата алюминия и аммония растворяют в 1 л дистиллированной воды, нагревают до 60° С и постепенно прибавляют 55 мл концентрированного раствора аммиака при постоянном перемешивании. После отстаивания в течение 1 ч осадок переносят в большой стакан и промывают декантацией дистиллированной водой до исчезновения свободного аммиака, хлоридов, нитритов и нитратов), тщательно перемешивают, оставляют стоять несколько минут и фильтруют. Первые порции фильтрата отбрасывают.
Окисление нитритов. К 100 мл пробы прибавляют 1 мл 1 н. раствора H2SO4 и перемешивают. Затем по каплям прибавляют при перемешивании необходимое количество 0,1 н. раствора перманганата калия или 3%-нога раствора перекиси водорода. Перманганат добавляют в таком количестве, чтобы розовая окраска не исчезала в течение не менее 15 мин. Поскольку окисление нитритов приводит к образованию нитратов, надо из результата определения последних вычесть количество нитритов, присутствовавших в первоначальной пробе (в пересчете на нитраты). Содержание нитритов определяют в отдельной порции пробы соответствующим методом.
Аппаратура
Фотометр с фиолетовым (синим) светофильтром ( ![]() = 410 нм). Кюветы с толщиной слоя 1-5 см или набор цилиндров Несслера емкостью 50 или 100 мл. Для повышения верхнего предела определяемой концентрации можно производить измерения и при других длинах волн, например 480 нм.
= 410 нм). Кюветы с толщиной слоя 1-5 см или набор цилиндров Несслера емкостью 50 или 100 мл. Для повышения верхнего предела определяемой концентрации можно производить измерения и при других длинах волн, например 480 нм.
Реактивы
Фенолдисульфоновая кислота, раствор в серной кислоте. Растворяют 25 г фенола ч. д. а, (препарат не должен быть окрашен) в 150 мл концентрированной серной кислоты. Прибавляют 75 мл дымящей серной кислоты (олеум с 15% SO3), тщательно перемешивают и нагревают с обратным холодильником в течение 2 ч на кипящей водяной бане.
Аммиак ч. д. а., концентрированный раствор.
Едкое кали, приблизительно 12 н. раствор. Растворяют 673 г КОН ч. д. а. в дистиллированной воде и доводят до 1 л.
Комплексон III, аммиачный раствор. Растирают 50 г комплексона III с 20 мл дистиллированной воды до получения пасты, которую растворяют в 50 мл концентрированного раствора аммиака.
Нитрат калия, стандартный раствор. Растворяют 0,1631 г KNOз ч. д. а., высушенного при 105°С, в дистиллированной воде, прибавляют 1 мл хлороформа и доводят водой до 1 л; 1 л содержит 0,100 м ![]() .
.
Калибровочная кривая. На водяной бане выпаривают досуха 50,0 мл стандарт кого раствора. Сухой остаток растворяют в 2,0 мл кислого раствора фенолсульфоновой кислоты и доводят дистиллированной водой до 100 мл.
Для визуального колориметрирования, проводимого в цилиндрах Несслера, в несколько цилиндров емкостью по 50 мл отмеривают 0,1; 0,3; 0,5; 0,7; 1,0, 2,0. 5,0; 10, 15; 20 и 30 мл окрашенного стандартного раствора (что отвечает 0; 0,1; 0,3; . . .I 30 мг ![]() в 1 л), прибавляют 2,0 мл раствора фенолдисульфоновой кислоты и такое же количество раствора NH4OH или КОН, какое прибавлялось в пробе.
в 1 л), прибавляют 2,0 мл раствора фенолдисульфоновой кислоты и такое же количество раствора NH4OH или КОН, какое прибавлялось в пробе.
Объем раствора в цилиндрах доводят дистиллированной водой до метки и перемешивают Приготовленные таким способом окрашенные стандарты для сравнение могут сохраняться несколько недель без изменения окраски.
Для построения калибровочного графика используют оптические плотности. окрашенных стандартов, приготовленных, как указано выше при описании визуального колориметрирования. Из найденных значений вычитают оптическую плотности холостой пробы. Строят график зависимости оптической плотности от концентрации нитрат-ионов.
Ход определения. Прозрачную пробу объемом 100 мл или менее с содержанием не более 5 мг ![]() нейтрализуют до рН 7, переливают в фарфоровую чашку и выпаривают на кипящей водяной бане досуха. К сухому остатку прибавляют 2,0 мл раствора фенолдисульфоновой кислоты и размешивают стеклянной палочкой до полного растворения. Если потребуется, смесь слегка нагревают на водяной бане. Прибавляют 20 мл дистиллированной воды и приливают при помешивании 6-7 мл концентрированного раствора аммиака или 5-6 мл 12 н. раствора едкого кали. Если при этом выделяются гидроокиси присутствующих металлов, их удаляют фильтрованием через стеклянный фильтр или прибавляют по каплям аммиачный раствор комплексона III до полного растворения осадка. Фильтрат или прозрачный раствор переносят в мерную колбу емкостью 50 или 100 мл, доводят дистиллированной водой до метки и содержимое колбы перемешивают. Окрашенный раствор колориметрируют. Из найденного значения вычитают оптическую плотность холостого раствора (дистиллированная вода с реактивами) и по калибровочному графику находят содержание нитрат-ионов.
нейтрализуют до рН 7, переливают в фарфоровую чашку и выпаривают на кипящей водяной бане досуха. К сухому остатку прибавляют 2,0 мл раствора фенолдисульфоновой кислоты и размешивают стеклянной палочкой до полного растворения. Если потребуется, смесь слегка нагревают на водяной бане. Прибавляют 20 мл дистиллированной воды и приливают при помешивании 6-7 мл концентрированного раствора аммиака или 5-6 мл 12 н. раствора едкого кали. Если при этом выделяются гидроокиси присутствующих металлов, их удаляют фильтрованием через стеклянный фильтр или прибавляют по каплям аммиачный раствор комплексона III до полного растворения осадка. Фильтрат или прозрачный раствор переносят в мерную колбу емкостью 50 или 100 мл, доводят дистиллированной водой до метки и содержимое колбы перемешивают. Окрашенный раствор колориметрируют. Из найденного значения вычитают оптическую плотность холостого раствора (дистиллированная вода с реактивами) и по калибровочному графику находят содержание нитрат-ионов.
Расчет. Содержание нитрат-ионов в мг/л (х) или в мг-екв/л (у) вычисляют по формулам:
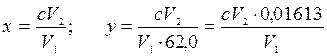
где с - концентрация нитрат-ионов, найденная по калибровочному графику или по шкале стандартов, мг/л; V1 - объем пробы, взятой для анализа, мл; V2 - объем окрашенной пробы, мл (50 или 100 мл); 62,0 - эквивалент ![]() .
.
Округление результатов
| Диапазон, мг/л | 0,10-1,00 | 1,0-2,0 | 2,0-5,0 | 5,0-10,0 |
| Округление ....... |
|
|
|
|
| в мг/л | 0,05 | 0,1 | 0,2 | 0,5 |
| в мг-экв/л | 0,001 | 0,002 | 0,005 | 0,01 |
Колориметрическое определение с салицилатом натрия
Определение основано на реакции нитратов с салицилатом натрия в среде серной кислоты, где образуются окрашенные в желтый цвет соли нитросалициловой кислоты. Без разбавления можно определять от 0,1 до 20 мг ![]() в 1 л воды.
в 1 л воды.
Мешающие влияния. Коллоидные органические и окрашенные вещества, присутствующие в пробе, мешают определению. Их удаляют так же, как и при определении нитратов с фенолдисульфоновон кислотой (см. выше).
Если сухой остаток после прибавления серной кислоты окрасится растворенными органическими веществами, то этот метод неприменим.
Определению мешают хлориды в количествах, превышающих 200 мг/л. Их удаляют способом, изложенным при определении нитратов с фенолдисульфоновой кислотой (см. там же).
Железо мешает определению в количестве, превышающем 5 мг/л. Мешающее влияние большой концентрации железа или других катионов можно устранить фильтрованием через катионит.
Нитриты оказывают влияние на определение нитратов при содержании их, превышающем 1-2 мг!л. Содержание 20 мг ![]() в 1 л повышает найденный результат при определении нитратов на 1 мг/л. Нитрит-ионы в количестве более 2 мг/л следует удалять выпариванием 20 мл пробы досуха на водяной бане с добавлением 0,05 г сульфата аммония.
в 1 л повышает найденный результат при определении нитратов на 1 мг/л. Нитрит-ионы в количестве более 2 мг/л следует удалять выпариванием 20 мл пробы досуха на водяной бане с добавлением 0,05 г сульфата аммония.
Аппаратура
Фотометр с фиолетовым светофильтром (![]() = 410 нм). Кюветы с толщиной слоя 1-5 см или набор цилиндров Несслера емкостью 50 мл. Водяная баня.
= 410 нм). Кюветы с толщиной слоя 1-5 см или набор цилиндров Несслера емкостью 50 мл. Водяная баня.
Реактивы.
Салицилат натрия, 0,5%-ный водный раствор (всегда свежеприготовленный).
Серная кислота ч. д. а., концентрированная, свободная от нитратов.
Едкий натр, приблизительно 10 н. раствор. Растворяют 400 г NaOH ч. д. а. в дистиллированной воде и после охлаждения доводят до 1 л.
Нитрат калия, стандартный раствор, содержащий в 1 мл 0,100 мг ![]() (приготовление - см. стр. 138).
(приготовление - см. стр. 138).
Рабочий раствор. Разбавляют 10,0 мл предыдущего раствора дистиллированной водой до 100 мл, всегда применяют свежеприготовленный раствор, 1 мл раствора содержит 0,010 мг ![]() .
.
Калибровочная кривая. Готовят стандарты, содержащие 0; 0,5; 1,0, 2,0. 5,0; 10,0; 20 мг ![]() в 1 л. Для этого берут 0; 0,5; 0,2 мл рабочего раствора нитрата калия и доводят дистиллированной водой до 10 мл, как описано ниже, затем строят калибровочный график в координатах оптическая плотность - концентрация нитрат-иона.
в 1 л. Для этого берут 0; 0,5; 0,2 мл рабочего раствора нитрата калия и доводят дистиллированной водой до 10 мл, как описано ниже, затем строят калибровочный график в координатах оптическая плотность - концентрация нитрат-иона.
Шкала стандартов. Для визуальной колориметрии в цилиндрах Несслера приготавливают окрашенные стандартные растворы объемом 50 мл, обработанные, как указано ниже, например, для ряда 0; 0,2; 0,5; 1,0; 2,0; 4.0* 6,0; 8,0; 10,0 мг ![]() в 1 л (0; 0,2; 0,5 и т. д. мл рабочего раствора доводят до 10 мл дистиллированной водой).
в 1 л (0; 0,2; 0,5 и т. д. мл рабочего раствора доводят до 10 мл дистиллированной водой).
Ход определения. К 10 мл пробы прибавляют 1 мл раствора салицилата натрия и выпаривают в фарфоровой чашке на водяной бане досуха. После охлаждения сухой остаток увлажняют 1 мл серной кислоты и оставляют на 10 мин. Содержимое чашки разбавляют дистиллированной водой, переносят количественно в мерную колбу емкостью 50 мл, прибавляют 7 мл 10 н. раствора едкого натра, доводят дистиллированной водой до метки и тщательно перемешивают. После охлаждения до комнатной температуры вновь доводят объем до метки и окрашенный раствор колориметрнруют. В течение 10 мин после прибавления раствора едкого натра окраска не изменяется. Из найденных значений оптической плотности вычитают оптическую плотность холостой пробы (приготовленной тем же способом с дистиллированной водой) и по калибровочному графику находят содержание нитрат-ионов.
Расчет. Расчет такой же, как и при определении нитратов с фе- нолдисульфоновой кислотой.
Округление результатов. Результаты округляют так же, как и в методе определения нитратов с фенолдисульфоновой кислотой.
Полярографическое определение
Нитраты восстанавливаются на ртутном капельном электроде в слабокислой среде при каталитическом действии ионов уранила. Фоном служит раствор хлорида калия и соляной кислоты, который содержит небольшое количество уранилацетата.
При полярографировании фона образуются две волны, первая из которых соответствует восстановлению урана (VI) до урана (V) с потенциалом полуволны -0,18 в по отношению к насыщенному каломельному электроду (НКЭ). Вторая волна соответствует восстановлению урана (V) до урана (IV) с потенциалом полуволны -0,94 в по отношению к НКЭ. В присутствии нитратов вторая волна повышается. При каталитическом характере электродной реакции повышение ее не пропорционально концентрации нитратов в растворе, однако в определенном диапазоне концентраций зависимость высоты волны от концентрации нитратов почти линейна.
Повышение второй полярографической волны урана, вызванное присутствием нитратов, зависит также от концентрации ионов уранила в рабочем электролите. С увеличением их концентрации полярографическая волна повышается. Границы линейной зависимости высоты волны от концентрации нитратов изменяются с изменением концентрации урана. Поэтому необходимо точно соблюдать Указанные в методике прописи концентрации.
Описанным методом можно определять не менее 1 мг ![]() в 1 л. Оптимальная концентрация от 5 до 30 мг
в 1 л. Оптимальная концентрация от 5 до 30 мг ![]() в 1 л воды.
в 1 л воды.
Мешающие влияния. Нитриты полярографически определяются так же, как и нитраты. Мешающее влияние нитритов можно не учитывать, если концентрация их незначительна (менее 0,2 мг/л), при более высоких концентрациях надо из полученного результата вычесть отдельно определенное содержание нитритов в пробе (1 мг ![]() равен 1,35 мг
равен 1,35 мг ![]() ).
).
Определению нитратов мешают также элементы, которые восстанавливаются на капельном ртутном электроде при потенциалах, близких к потенциалу полуволны второй волны урана. Оказывают влияние элементы, которые восстанавливаются при более положительных потенциалах.
Повышенное содержание органических веществ оказывает неблагоприятное влияние на форму полярографической волны, сокращает верхний участок кривой и не дает возможности точно измерить высоту волны. Поэтому нельзя полярографически определять нитраты в пробах сточных или сильно загрязненных природных вод, содержащих большие количества органических веществ.
Пробы, имеющие щелочную или сильнокислую реакцию, перед определением необходимо нейтрализовать 1 н. раствором едкого натра или соляной кислотой по метиловому оранжевому. Присутствие его не мешает определению. При расчете необходимо учитывать изменение объема пробы.
Ошибки, которые могут возникнуть вследствие неодинаковой концентрации ионов уранила в исследуемом и в стандартных растворах, необходимо заранее исключить путем очень точного измерения раствора фона и пробы.
Аппаратура
Полярограф.
Полярографические ячейки.
Баллон со сжатым азотом.
Промывалки для удаления следов кислорода из азота, наполненные амальгамой цинка и раствором соли хрома (III) в соляной кислоте или насыщенным раствором ![]() -антрахинонсульфоната натрия в 10%-ном растворе едкого натра.
-антрахинонсульфоната натрия в 10%-ном растворе едкого натра.
Реактивы
Фон. Растворяют 37,28 г. хлорида калия ч. д. а, в 400 мл дистиллированной воды, приливают 5 мл концентрированной соляной кислоты ч. д. а. и прибавляют 0,212 г уранилацетата UO2(CH3COO)2·2H2О ч. д. а.; раствор доводят дистиллированной водой до 500 мл.
Нитрат, калия, стандартный раствор, содержащий в 1 мл 0,100 мг ![]() (приготовление см. «Колориметрическое определение с фенолдисульфоновой кислотой»).
(приготовление см. «Колориметрическое определение с фенолдисульфоновой кислотой»).
В мерную колбу емкостью 1 л помещают 300 мл приготовленного раствора и доводят дистиллированной водой до метки. В 1 мл раствора содержится 0,030 мг ![]() . Для построения калибровочного графика следует применять свежеприготовленный раствор.
. Для построения калибровочного графика следует применять свежеприготовленный раствор.
Калибровочная кривая. Для построения калибровочного графика используют стандартные растворы с концентрациями 0; 1,0; 3,0; 6,0; 9,0; . .; 27,0; 30,0 мг ![]() в 1 л 0; 3,33, 10; 20; 30; . .; 90-100 мл. Для этого рабочий стандартный раствор разбацляют до 100 мл дистиллированной водой и обрабатывают так же, как при анализе пробы.
в 1 л 0; 3,33, 10; 20; 30; . .; 90-100 мл. Для этого рабочий стандартный раствор разбацляют до 100 мл дистиллированной водой и обрабатывают так же, как при анализе пробы.
Строят два калибровочных графика. Один -для концентраций от 0 до 12 мг ![]() в 1 д, другой - для концентраций от 0 до 30 мг
в 1 д, другой - для концентраций от 0 до 30 мг ![]() в 1 л. Чувствительность выбирают так, чтобы при высотах волн, изморенных для наиболее концентрированных растворов обоих рядов (12 и 30 мг!л), была максимально использована высота регистрационной бумаги.
в 1 л. Чувствительность выбирают так, чтобы при высотах волн, изморенных для наиболее концентрированных растворов обоих рядов (12 и 30 мг!л), была максимально использована высота регистрационной бумаги.
Графики периодически проверяют (анализируя стандартные растворы) и строят новые графики для нового фона.
Ход определения. В полярографическую ячейку отмеривают пипеткой 15,0 мл или меньший объем пробы, доведенный до 15,0 мл дистиллированной водой, и при помощи микробюретки прибавляют 1,00 мл фона. Растворенный «недород удаляют, продувая раствор азотом. Полярографическую кривую строят, начиная от значений потенциалов -0,6 в по отношению к донной ртути.
Чувствительность, высота ртутного столба и способ измерения высоты волны должны быть такими же, как и при калибровании электрода. Отклонение от температуры калибрования допускается в пределах ±2° С. По найденной высоте полуволны, пользуясь калибровочным графиком, определяют содержание нитрат-ионов.
Расчет. Содержание нитрат-ионов в мг!л (х) и в мг-екв/А (у) вычисляют по формулам:
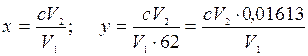
где с - концентрация нитратов, найденная по калибровочному графику, мг/л; V1 - объем первоначальной пробы, взятой для разбавления, мл; V2 - объем пробы после разбавления, мл; 62 - эквивалент ![]() .
.
Округление результатов
| Диапазон, мг/л | 1,0-10,0 | 10,0-20,0 | 20-50 | 50-100 |
| Округление ....... |
|
|
|
|
| в мг/л | 0,2 | 0,5 | 1 | 2 |
| в мг-экв/л | 0,005 | 0,01 | 0,02 | 0,05 |
Определение восстановлением нитрат-ионов до аммиака
Определение основано на восстановлении нитратов до аммиака водородом в момент выделения, образующимся при реакции едкого кали со сплавом Деварда. Аммиак отгоняют из реакционной смеси и улавливают в приемнике с серной кислотой, в котором его затем определяют либо объемным методом, либо колориметрически. Колориметрический метод следует применять при концентрации нитратов ниже 10 мг/л, при концентрации выше 5 мг/л ![]() определение проводят объемным методом.
определение проводят объемным методом.
Мешающие влияния. Результаты определения искажаются в присутствии аммиака и нитритов. Аммиак или образующие его в условиях определения вещества устраняют выпариванием щелочной пробы, как описано в ходе определения. Нитриты необходимо определить отдельно и полученный результат вычесть из результата определения нитратов.
Аппаратура
Перегонный аппарат.
Аппаратура для определения аммиака.
Реактивы
Едкий натр, 25%-пни раствор. Растворяют 250 в NaOH ч.д. а, в 1250 мл дистиллированной воды, прибавляют около 0,5 г сплава Деварда и упариванием доводят объем до 1 л.
Сплав Деварда (порошок).
Бидистиллят, свободный от аммиака.
Реактив Несслера.
Серная кислота, 0,02 и, раствор.
Едкий, цитр, 0,02 н. раствор.
Смешанный индикатор (приготовление - см. выше).
Ход определения. К 200 мл пробы, налитой в перегонную колбу, прибавляют 20 мл 25%-наго раствора едкого натра и выпаривают смесь до половины ее объема. После охлаждения прибавляют 200 мл бидистиллята и около 0,5 г сплава Деварда. Колбу быстро соединяют изогнутой насадкой с холодильником. Конец холодильника должен быть погружен в 0,02 н. раствор серной кислоты, находящейся в приемнике. Смесь в перегонной колбе сначала нагревают слабо и только после окончания выделения водорода доводят до кипения. После отгонки около 150 мл жидкости перегонку прекращают. Содержимое приемника разбавляют бидистиллятом до 200 мл.
Обработку содержимого в приемнике и определение аммиака в дистилляте (колориметрически или титрованием) проводят так же, как при определении аммиака. Одновременно с анализом пробы определяют количество титрованного раствора, расходуемого на холостой опыт с бидистиллятом.
Расчет. Содержание нитрат-ионов при колориметрическом определении в мг1л (х) или в мг-экв!л (у) вычисляют по формулам:

где а - концентрация аммиака, выраженная в мг NH+4/л; b - результат отдельно проведенного определения нитритов, выраженный в мг ![]() /л, 3,44 - коэффициент пересчета NH+4 на
/л, 3,44 - коэффициент пересчета NH+4 на ![]() ; 1,35 - коэффициент пересчета
; 1,35 - коэффициент пересчета ![]() на
на ![]() ; 62- эквивалент
; 62- эквивалент ![]() .
.
Содержание нитрат-ионов при определении объемным методом в мг/л (х) или в мг-екв/л (у) вычисляют по формулам:
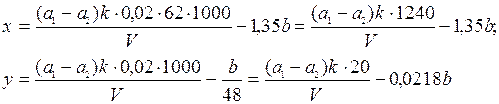
где a1 - объем 0,02 н. раствора едкого натра, израсходованного на холостой опыт при титровании 25,0 мл 0,02 н. раствора серной кислоты, м; a2 - объем 0,02 н. раствора едкого натра, израсходованного на титрование пробы, мл; V - объем пробы, взятой для определения, мл; k - поправочный коэффициент для приведения концентрации раствора NaOH к точно 0,02 н.; b - значение приведено выше.
Округление результатов
| Диапазон, мг/л | 2,0-5,0 | 5,0-10,0 | 10-20 | 20-50 |
| Округление ....... |
|
|
|
|
| в мг/л | 0,2 | 0,5 | 1 | 2 |
| в мг-экв/л | 0,05 | 0,01 | 0,02 | 0,05 |
Колориметрическое определение с бруцином
Нитраты реагируют с бруцином в сернокислой среде с образованием соединений, цвет которых изменяется от желтого до коричнево- красного. Интенсивность возникающей окраски не вполне подчиняется закону Бора. Для каждой серии проб строят калибровочный график. Без разбавления нитраты определяются при концентрациях 1-20 мг ![]() в 1 л воды (0,016-0,323 мг-екв/л). Точность определения ±0,5 мг/л.
в 1 л воды (0,016-0,323 мг-екв/л). Точность определения ±0,5 мг/л.
Мешающие влияния. Определению мешают все сильные окислители и восстановители. Присутствие некоторых окислителей может быть обнаружено о-толидином, как при определении свободного хлора. Мешающее влияние остаточного хлора в концентрациях До 5 мг/л можно устранить добавлением арсенита натрия (0,1 мг 0,183%-нога раствора Na3AsO3 на 0,05 мг Cl2). Небольшой избыток арсенита не оказывает мешающего влияния.
Соли двух- и трехвалентного железа, а также соединения марганца (IV) мешают при их концентрациях выше 1 мг1л. Мешающее влияние нитритов устраняется сульфаниловой кислотой, содержащейся в бруциновом реактиве. Хлориды определению не мешают.
Интенсивность возникающей окраски и скорость ее возникновения в значительной мере зависят от температуры, времени и действия света. Для достижения надежных и воспроизводимых результатов необходимо поступать точно в соответствии с методикой (стаканы всегда емкостью 50 мл и т. д.). Поскольку для определения нужно очень малое количество пробы, следует брать точный объем 66 и точно проводить разбавление.
Аппаратура
Стаканы емкостью 50 мл, одинакового диаметра - 30 штук.
Фотометр с фиолетовым светофильтром (![]() = 410 нм). Кюветы с толщиной слоя более 2 см.
= 410 нм). Кюветы с толщиной слоя более 2 см.
Реактивы
Бруциновый реактив. Растворяют 1 г бруцина и 0,1 г сульфаниловой кислоты в 70 мл горячей дистиллированной воды, прибавляют около 3 мл концентрированной соляной кислоты и объем.доводят дистиллированной водой до 100 мл. Раствор сохраняется в течение 2 месяцев. Слабо-розовая окраска, которая проявляется через некоторое время, не мешает. Бруцин - сильный яд/ Необходимо соблюдать осторожность при отмеривании пипеткой или взвешивании.
Серная кислота, 90%-ный раствор. Осторожно при перемешивании прибавляют к 75 мл дистиллированной воды 500 мл концентрированной серной кислоты. Перед употреблением раствор охлаждают до комнатной температуры.
Серная кислота не должна содержать нитратов. В случае их присутствия они устраняются следующим образом. К серной кислоте прибавляют несколько кристаллов сульфата аммония (лучше всего в большой колбе Кьельдаля) и нагревают не менее 1 ч при температуре около 300° С.
Нитрат калия, стандартный раствор, содержащий 0,100 мг Nog в 1 мл (приготовление - см. выше).
Рабочий раствор. Разбавляют 50,0 мл приготовленного раствора дистиллированной водой до 250 мл. В 1 мл раствора содержится 0,020 мг ![]() .
.
Ход определения. В пять мерных колб емкостью 100 мл вводят пипеткой 0; 25; 50; 75 и 100 мл рабочего стандартного раствора и доводят дистиллированной водой до метки. Концентрация этих стандартов 5; 10; 15 и 20 мг ![]() в 1 д.
в 1 д.
В первые пять стаканов емкостью 50 мл помещают пипеткой точно по 2,00 мл приготовленного раствора. В другие пять - по 2,00 мл пробы, содержащей от 1 до 20 мг NOa в 1 л (одновременно можно анализировать не более 10 проб). К стандартам и пробам прибавляют по 1,0 мл бруцинового реактива.
Во второй ряд стаканов емкостью 50 мл отмеривают (лучше из автоматической бюретки) по 10 мл 90%-ной серной кислоты. После этого раствор из первого стакана переливают в соответствующий ему стакан с кислотой. Для полного смешения жидкость переливают из стакана в стакан от трех до пяти раз. Этот процесс повторяют со всеми приготовленными стандартными растворами и пробами. Число переливаний должно быть для всего ряда растворов одинаковым, и вся операция должна быть проведена быстро. Стаканы со стандартными растворами и пробами ставят в темное место на 10 ± 1 мин для возникновения окраски. В оставшийся ряд пустых стаканов отмеривают по 10 мл дистиллированной воды.
Содержание нитратов находят по калибровочному графику, который строят по результатам измерений оптической плотности четырех стандартных растворов, за вычетом оптической плотности холостой пробы.
Округление результатов
| Диапазон, мг/л | 1,0-20,0 | 20-50 | 50-100 | 100-200 |
| Округление ....... |
|
|
|
|
| в мг/л | 0,5 | 2 | 5 | 10 |
| в мг-экв/л | 0,01 | 0,05 | 0,10 | 0,2 |
Похожие работы
... разовая) – 0,01%. 4 Содержание Введение......................................................................................................................4 Глава 1. Межпредметные связи в курсе школьного предмета химии на примере углерода и его соединений.......................................................................5 1.1 Использование межпредметных связей для формирования у учащихся ...
... в процессе его производства. Первая стадия производства титана заключается в рудно-восстановительной плавке, которая проводится с целью обогащения исходного материала окисными соединениями титана. Во всех последующих стадиях производства взаимодействие титана и его соединений с кислородом нежелательно. Титан растворяет такие элементы, как азот, водород и углерод. С последним он образует стойкие ...
... среды. 3.1 Урок по теме «Кальций и его соединения» в 9-ом классе с. Карасу, позволяющий развить экологическое сознание школьников Цель урока: познакомить учащихся с основными способами получения кальция и его соединений, возможностями применения соединений кальция, показать необходимость контроля содержания ионов кальция и магния в питьевых и сточных водах и обозначить значимость кальция ...
... разделам школьной программе по химии, тем более, что в учебнике этот материал, по-моему, незаслуженно отсутствует. Данная работа посвящена изучению основных физических и химических свойств хрома и его соединений, позволяет оценить важность этого химического элемента. 1.Исторические сведения В 1766 году петербургский профессор химии И.Г.Леман описал новый минерал, найденный на Урале на ...
0 комментариев