Ковалентная
(неполярная,
полярная) связь.
Механизмы
образования
ковалентной
связи
Году американский
ученый Льюис
высказал
предположение
о том, что химическая
связь образуется
за счет обобществления
двух электронов
Скорость
химических
реакций. Порядок
и молекулярность
реакций
Зависимость
скорости реакции
от температуры.
Правило Вант-Гоффа.
Уравнение
Аррениуса
Энергия
активации.
Активированные
комплексы.
Уравнение
Аррениуса
Растворы.
Физическая
и химическая
теории растворов
Фазовые
равновесия
в гетерогенных
системах, фазовые
превращения
и правило фаз.
Диаграммы
состояния
Сильные
и слабые электролиты.
Константа
диссоциации.
Закон разбавления
Оствальда.
Слабые электролиты.
Константа
диссоциации
Электролитическая
диссоциация
воды. Ионное
произведение
воды. Водородный
показатель
среды. Понятие
об индикаторах
Гидролиз
солей. Обратимый
и необратимый
(полный) гидролиз.
Роль процессов
гидролиза при
эксплуатации
котельных
установок.ъ
Растворимость
веществ. Произведение
растворимости.
Механизм
накипеообразования
Концентрации
или парциальные
давления окисленной
и восстановленной
форм
Окислитель
или восстановитель
иногда дополнительно
расходуется
на связывание
получающихся
продуктов
(солеобразование).Например,
в реакции
Электродный
потенциал.
Влияние температуры
и концентрации
на величину
электродного
потенциала.
Уравнение
Нернста
Практическое
использование
электрохимических
процессов.
Химические
источники тока
Коррозия
металлов. Основные
виды коррозии.
Химическая
коррозия
Коррозия
металлов.
Электрохимическая
коррозия
Методы защиты
металлов от
коррозии: изменение
свойств коррозионной
среды, защитные
покрытия,
электрохимическая
защита
Распространенность
химических
элементов.
Основные классы
неорганических
соединений
Навигация
Году американский ученый Льюис высказал предположение о том, что химическая связь образуется за счет обобществления двух электронов
Общая и неорганическая химия
442397
знаков
6
таблиц
13
изображений
1916 году американский ученый Льюис высказал предположение о том, что химическая связь образуется за счет обобществления двух электронов.
При этом электронная оболочка атома стремится по строению к электронной оболочке благородного газа. В дальнейшем эти предположения послужили основой для развития метода валентных связей. В 1927году Гайтлером и Лондоном был выполнен теоретический расчет энергии двух атомов водорода в зависимости от расстояния между ними. Оказалось, что результаты расчета зависят от того, одинаковы или противоположны по знаку спины взаимодействующих электронов. При совпадающем направлении спинов сближение атомов приводит к непрерывному возрастанию энергии системы. При противоположно направленных спинах на энергетической кривой имеется минимум, т.е. образуется устойчивая система – молекула водорода Н2.
Образование химической связи между атомами водорода является результатом взаимопроникновения (перекрывания) электронных облаков. Вследствие этого перекрывания плотность отрицательного заряда в межъядерном пространстве возрастает, и положительно заряженные ядра притягиваются к этой области.
Представления о механизме образования молекулы водорода были распространены на более сложные молекулы. Разработанная на этой основе теория химической связи получила название метод валентных связей (метод ВС).
1) Ковалентная связь образуется двумя электронами с противоположно направленными спинами, причем эта электронная пара принадлежит двум атомам.
2) Ковалентная связь тем прочнее, чем в большей степени перекрываются электронные облака.
Комбинации двухэлектронных двухцентровых связей, отражающие электронную структуру молекулы, получили название валентных схем. Примеры построения валентных схем:
![]()
![]()
Ковалентная связь характеризуется длиной, энергией, полярностью, поляризуемостью и имеет определённую направленность в пространстве.
С увеличением
кратности
связи, длина
связи уменьшается: ![]() −
−![]() 0,154 нм
0,154 нм ![]() =
=![]() 0,134 нм
0,134 нм![]() ≡
≡![]() 0,12 нм
0,12 нм
Энергия связи – энергия, которую надо затратить, чтобы разорвать химическую связь. Тоже количество энергии выделяется при образовании химической связи. С увеличением кратности связи, энергия увеличивается. Энергия p-связи меньше энергии s-связи.
Свойства ковалентной связи: прочность, полярность, насыщаемость, направленность, гибридизация, кратность
1. Прочность ковалентной связи — это свойства характер длинной связи (межъядерное пространство) и энергии энергией связи.
2. Полярность ковалентной связи. В молекулах, содержащих ядра атомов одного и того же элемента, одна или несколько пар электронов в равной мере принадлежат обоим атомам, каждое ядро атома с одинаковой силой притягивает пару связывающих электронов. Такая связь называется неполярной ковалентной связью. Если пара электронов, образующих химическую связь, смещена к одному из ядер атомов, то связь называют полярной ковалентной связью.
3. Насыщаемость ковалентной связи — это способность атома участвовать только в определенном числе ковалентной связи, насыщаемость характеризует валентностью атома. Количественные меры валентности являются число не спаренных электронов у атома в основном и в возбужденном состоянии.
4. Направленность ковалентной связи. Наиболее прочные ковалентной связи образуются в направлении максимального перекрывания атомных орбиталей, то есть мерой направленности служит валентный угол.
5. Гибридизация ковалентной связи — при гибридизации происходит смещение атомных орбиталей, т.е. происходит выравнивание по энергии и по форме. Существует sp, sp2, sp3 —гибридизация. sp — форма молекулы линейная (угол 1800), sp2 — форма молекулы плоская треугольная (угол 1200), sp3 - форма тетраэдрическая (угол 109028).
6. Кратность ковалентной связи или делоколизация связи — Число связей, образующихся между атомами, называется кратностью (порядком) связи. С увеличением кратности (порядка) связи изменяется длина связи и ее энергия.
Типы химической связи. Ионная, металлическая связи
Металлическая связь При обычных условиях металлы, за исключением ртути Hg, существуют в виде кристаллов. Взаимодействие, удерживающее атомы металлов в едином кристалле, называется металлической связью.
Природа металлической связи подобна ковалентной связи: оба типа связи основаны на обобществлении валентных электронов. Однако в атомах металлов количество таких электронов меньше количества вакантных орбиталей. Электроны слабо удерживаются ядром. Поэтому они могут переходить из одной орбитали в другую. Стремясь принять более устойчивое состояние, а это структура инертного газа, атомы металлов довольно легко отдают валентные электронные электроны, превращаясь в положительно заряжённые ионы. Внутри этой решётки находятся валентные электроны, которые не принадлежат конкретно какому-то атому. Благодаря малым размерам электроны более или менее свободно перемещаются по всему объёму кристаллической решётки, поэтому возникает большое число многоцентрированных орбиталей. Электроны на этих орбиталях обобщены сразу несколькими атомами.
Благодаря свободному перемещению электронов металлы обладают высокой электрической проводимостью и теплопроводностью.
По прочности
металлическая
связь меньше
ковалентной
связи в 3-4 раза.
Металлическая
связь не имеет
определённой
направленности
в пространстве.
Электроны
сталкиваясь
с ионами образуют
нейтральные
частицы, которые
сразу теряют
электроны: ![]() .
Электронные
газы отражают
световые лучи.
.
Электронные
газы отражают
световые лучи.
В результате движения внутри решётки электроны способны переносить тепловую энергию от нагретых участков к ненагретым, этим объясняется теплопроводность.
Если приложить нагрузку к металлу, происходит деформация без разрушения решётки, металлам характерна ковкость, пластичность.
Ионная Химическая связь, осуществляемая за счет электростатического притяжения между ионами, называется ионной связью. Соединения, образованные путем притяжения ионов называются ионными. Ионные соединения состоят из отдельных молекул только в парообразном состоянии. В твердом (кристаллическом) состоянии ионные соединения состоят из закономерно расположенных положительных и отрицательных ионов. Молекулы в этом случае отсутствуют. Ионные соединения образуют резко различные по величине электроотрицательности элементы главных подгрупп I и II групп и главных подгрупп VI и VII групп. В зависимости от величины электроотрицательности все элементы делятся на:
электроположительные (элементы 1-3 группы)
электротрицательные (все остальные элементы)
Ионная связь образуется между элементами сильно отличающимися по электроотрицательности.
Ионных соединений сравнительно немного. Например неорганические соли: NH4Cl (ион аммония NH4 + и ион хлора Cl-), а также солеобразные органические соединения: алкоголяты соли карбоновых кислот, соли аминов Неполярная ковалентная связь и ионная связь — два предельных случая распределения электронной плотности.
Неполярной связи отвечает равномерное распределение связующего двух электронного облака между одинаковыми атомами. Наоборот, при ионной связи связующие электронное облако практически полностью принадлежит одному из атомов.
В большинстве же соединений химические связи оказывают промежуточными между этими видами связи, то есть в них осуществляется полярная ковалентная связь.
Потенциал ионизации – энергия, которую необходимо затратить для удаления 1-го электрона с внешней орбитали, при этом атом переходит из нейтрального в положительно заряженный ион (катион).
Чем меньше потенциал ионизации, тем легче атом теряет электроны, тем сильнее выражены у электрона металлические свойства. Потенциал ионизации растет в пределах периода слева направо, уменьшается сверху вниз.
Ионная связь образуется за счет перехода одного или нескольких электронов от одного атома на внешнюю оболочку другого атома.
Атом, отдавший электрон становится положительно заряженным, а получивший – отрицательно заряженный Связь между разноименными ионами осуществляется за счет сил электростатического притяжения.
Образование ионной связи происходит по октаэдрическому правилу. Согласно этому правилу атом принимает, теряет или разделяет электроны таким образом, чтобы электронное облако для него соответствовало ближайшему инертному газу.
Основные виды взаимодействия молекул. Силы межмолекулярного взаимодействия. Водородная связь
Межмолекулярные взаимодействия, взаимодействия молекул между собой, не приводящее к разрыву или образованию новых химических связей. Межмолекулярные взаимодействия определяют отличие реальных газов от идеальных, существование жидкостей и молекулярных кристаллов. От межмолекулярных взаимодействий зависят многие структурные, спектральные, термодинамические, теплофизические и другие свойства веществ. Появление понятия межмолекулярные взаимодействия связано с именем Й. Д. Ван-дер-Ваальса, который для объяснения свойств реальных газов и жидкостей предложил в 1873 уравнение состояния, учитывающее межмолекулярные взаимодействия. Поэтому силы межмолекулярного взаимодействия часто называют ван-дер-ваальсовыми.
Виды межмолекулярных взаимодействийОснову межмолекулярных взаимодействий составляют кулоновские силы взаимодействия между электронами и ядрами одной молекулы и ядрами и электронами другой. В экспериментально определяемых свойствах вещества проявляется усредненное взаимодействие, которое зависит от расстояния R между молекулами, их взаимной ориентации, строения и физических характеристик (дипольного момента, поляризуемости и др.). При больших R, значительно превосходящих линейные размеры l самих молекул, вследствие чего электронные оболочки молекул не перекрываются, силы межмолекулярного взаимодействия можно достаточно обоснованно подразделить на три вида - электростатические, поляризационные (индукционные) и дисперсионные. Электростатические силы иногда называют ориентационными, однако это неточно, поскольку взаимная ориентация молекул может обусловливаться также и поляризационными силами, если молекулы анизотропны.
При малых расстояниях между молекулами (R ~ l) различать отдельные виды межмолекулярных взаимодействий можно лишь приближенно, при этом, помимо названных трех видов, выделяют еще два, связанные с перекрыванием электронных оболочек, - обменное взаимодействие и взаимодействия, обязанные переносу электронного заряда. Несмотря на некоторую условность, такое деление в каждом конкретном случае позволяет объяснять природу межмолекулярного взаимодействия и рассчитать его энергию.
Водородная связь Водородные связи могут образовываться между атомом водорода, связанным с атомом электроотрицательного элемента, и электроотрицательным элементом, имеющим свободную пару электронов(О,F,N). Водородная связь обусловлена электростатическим притяжением, которому способствуют малые размеры атома водорода, и отчасти, донорно-акцепторным взаимодействием. Водородная связь может быть межмолекулярной и внутримолекулярной. Связи 0-Н имеют выраженный полярный характер: Водородная связь гораздо более слабая, чем ионная или ковалентная, но более сильная, чем межмолекулярное взаимодействие. Водородные связи обуславливают некоторые физические свойства веществ (например, высокие температуры кипения). Особенно распространены водородные связи в молекулах белков, нуклеиновых кислот и других биологически важных соединений, обеспечивая им определенную пространственную структуру (организацию).
В 80-х годах XIX в. М.А. Ильинский и Н.Н. Бекетов установили, что атом водорода, соединенный с атомом фтора, кислорода или азота, способен образовывать еще одну дополнительную связь – то есть некоторые водородосодер-жащие группы атомов образуют химическую связь, электроотрицательные атомы которой входят в состав молекулы. Этот вид связи получил название водородная связь.
Водородная связь – взаимодействие между двумя электроотрицательными атомами одной или нескольких разных молекул при помощи атома водорода: А—Н...В (чертой обозначена ковалентная связь, тремя точками – водородная связь).Для водородной связи характерно электростатическое притяжение водорода (несущего положительный заряд ?+) к атому электроотрицательного элемента, имеющего отрицательный заряд ?-. Чаще всего она слабее ковалентной, но сильнее обычного притяжения молекул друг к другу в твердых и жидких веществах.Водородная связь отличается от межмолекулярных взаимодействий тем, что обладает свойствами направленности и насыщаемости.Водородная связь считается разновидностью ковалентной химической связи. Описывается при помощи метода молекулярных орбита-лей в виде трехцентровой двухэлектронной связи.Признак наличия водородной связи – расстояние между атомом водорода и другим атомом, ее образующим, меньше, чем общая сумма радиусов этих атомов.
Чаще встречаются несимметричные водородные связи (расстояние Н...В>А—В ), редко – симметричные (HF ).Угол между атомами А—Н...В ~180o.Водородная связь присутствует во многих химических соединениях. Образуется между наиболее электроотрицательными элементами (фтор, азот, кислород), реже – в некоторых других (хлор, сера).Наиболее прочные водородные связи имеются в воде, фтороводороде, кислородсодержащих неорганических кислотах, карбоновых кислотах, фенолах, спиртах, аммиаке, аминах.
При кристаллизации водородные связи сохраняются. Кристаллические решетки водородных связей:
1) цепи (метанол);2) плоские двухмерные слои (борная кислота);3) пространственные трехмерные сетки (лед).Внутримолекулярная водородная связь – водородная связь, объединяющая части одной молекулы.
Межмолекулярная водородная связь – водородная связь, образующаяся между атомом водорода одной молекулы и атомом неметалла другой молекулы.
Цепные реакции. Механизм протекания цепных реакций
Существуют химические реакции, в которых взаимодействие между компонентами происходит довольно просто. Существует весьма обширная группа реакций, протекающих сложно. В этих реакциях каждый элементарный этап связан с предыдущим, без выполнения которого дальнейшая реакция невозможна. В таких реакциях образование продукта реакции являет собой результат цепи элементарных этапов реакции, что называется цепными реакциями, которые проходят при участии активных центров – атомов, ионов или радикалов (осколков молекул). Радикал – осколок молекулы, имеющий неспаренные электроны и проявляющий высокую реакционную активность (H, Cl, O, OH, CH3).
При взаимодействии активных центров с молекулами исходных компонентов происходит образование продуктов реакции и новых активных частиц, способствующих новому этапу взаимодействия. Активные центры способствуют и создают цепи последовательных превращений веществ.
В качестве примера цепной реакции можно привести реакцию синтеза хлористого водорода:
Эту реакцию провоцирует свет. Молекула хлора поглощает квант лучистой энергии hv и приходит в возбуждение, то есть атом в ней начинает энергично колебаться. Когда энергия колебаний превышает энергию связи, то происходит распад молекулы (фотохимическая диссоциация ):
Обрыв цепи – окончание цепи, характеризующееся соударением двух активных частиц и одной неактивной, результатом которой является образование молекулы и унос выделившейся энергии неактивной частицей.Цепные реакции делятся на: 1) неразветвленные цепные реакции;2) разветвленные цепные реакции.Неразветвленная цепная реакция характеризуется тем, что при каждом элементарном взаимодействии один активный центр образует молекулу продукта реакции и один новый активный центр. Разветвленная цепная реакция характеризуется тем, что по ходу взаимодействия свободного радикала с молекулой исходного реагента происходит образование нескольких новых активных центров, одни из которых дают начало новым активным центрам, а другие продолжают старую цепь.
Пример разветвленной цепной реакции – реакция образования воды из простых веществ:
Теория разветвленных цепных реакций была выдвинута Н.Н. Семеновым в 20-х годах XX века при изучении кинетики разнообразных процессов. Теория цепных реакций является научной основой для отраслей техники. Ядерные цепные реакции тоже относятся к цепным процессам.
ЦЕПНЫЕ РЕАКЦИИ – химические реакции, идущие путем последовательности одних и тех же элементарных стадий, на каждой из которых возникает одна или несколько активных частиц (атомов, свободных радикалов, ионов, ион-радикалов). По цепному механизму протекают реакции крекинга, горения, полимеризации и ряд других реакций
Цепи Боденштейна – Нернста. К концу 19 в. была разработана важнейшая глава физической химии – учение о равновесиях химических реакций (химическая термодинамика). Стало возможным рассчитывать, на какую максимально возможную глубину может пройти конкретная реакция при данных условиях. Одновременно создавалось учение о скоростях химических процессов – химическая кинетика. Накопленные ко второй половине 19 в. многочисленные экспериментальные данные можно было объяснить на основании закона действующих масс и уравнения Аррениуса. В то же время появлялись факты, которые невозможно было объяснить ни одной из существовавших теорий. Одной из самых загадочных оказалась очень простая с виду реакция водорода с хлором: H2 + Cl2 ® 2HCl.
В 1845 английский химик Джон Дрепер обнаружил, что под действием солнечного света хлор приобретает особую активность в реакции с водородом (см. ФОТОХИМИЯ). Еще более удивительный факт обнаружили в 1857 немецкий химик Роберт Бунзен и его ученик из Англии Генри Роско. Оказалось, что некоторые примеси даже в самых малых концентрациях могут оказать огромное влияние на скорость этой реакции. Например, малые добавки кислорода замедляли ее в сотни раз. Это был парадоксальный результат, так как кислород сам прекрасно реагирует с водородом. Обнаружились и другие непонятные явления. Например, скорость реакции зависела от материала стенки сосуда и даже от его размеров. В стройном, казалось бы, учении о скоростях реакций появилась брешь, и никто не знал, как с ней справиться.
А реакция водорода с хлором преподносила ученым все новые сюрпризы. В начале 20 в. Альберт Эйнштейн сформулировал закон, согласно которому каждый поглощенный квант света (фотон) вызывает изменения лишь в одной молекуле. Экспериментально несложно измерить число прореагировавших (или образовавшихся) молекул и число поглощенных в реакции квантов света. Отношение этих величин называется квантовым выходом реакции. Так, если на каждый поглощенный реагентами квант света образуется одна молекула продукта, то квантовый выход такой реакции равен единице. Однако экспериментально измеренные квантовые выходы многих реакций не соответствовали закону квантовой эквивалентности. В 1913 один из основоположников химической кинетики немецкий химик Макс Боденштейн измерил квантовый выход фотохимической реакции водорода с хлором H2 + Cl2 ® 2HCl. Результат оказался невероятным: число молекул HCl, образовавшихся при поглощении смесью одного кванта света, в некоторых условиях достигал миллиона! Боденштейн объяснил этот поразительный результат единственным разумным методом: каждый поглощенный квант света «запускает» длинную цепочку превращений, в которой реагируют сотни тысяч молекул исходных веществ (H2 и Cl2), превращаясь в молекулы продукта реакции (HCl). Это похоже на то, как выстроенные в ряд костяшки домино быстро, как по команде, падают одна за другой, если удачно толкнуть первую из них.
Боденштейном были сформулированы и основные принципы протекания нового типа химических превращений – цепных реакций. Эти реакции обязательно имеют три стадии: 1) зарождение цепи, когда происходит образование активных частиц; 2) продолжение (развитие) цепи; 3) обрыв цепи. Зарождение цепей в тепловой реакции происходит в результате диссоциации молекул при нагревании. В фотохимической реакции зарождение цепей происходит при поглощении кванта света. На стадии продолжения цепи образуются молекулы продуктов реакции и одновременно появляется новая активная частица, способная продолжать цепь. На стадии обрыва происходит исчезновение (дезактивация) активной частицы.
При сильном нагреве или при интенсивном освещении ультрафиолетовым светом цепная реакция водорода с хлором идет со взрывом. Но если температура не очень высокая или интенсивность света невелика, реакция идет спокойно. Основываясь на этом факте, Боденштейн выдвинул очень важный принцип стационарной концентрации промежуточных продуктов цепных реакций. В соответствии с этим принципом, скорость генерирования активных частиц на стадии зарождения равна скорости их исчезновения на стадии обрыва. Действительно, если бы скорость обрыва была больше скорости зарождения цепей, число активных частиц снизилось бы до нуля, и реакция прекратилась бы сама собой. В случае же преобладания скорости зарождения, число активных частиц росло бы со временем, что привело бы к взрыву.
Однако выяснение химического механизма для каждой стадии реакции водорода с хлором оказалось трудной задачей. Боденштейн предположил теорию энергетического разветвления: образующиеся в первичной реакции молекулы HCl несут избыточную энергию и потому способствуют протеканию дальнейших реакций, передавая избыток энергии молекулам исходных веществ. Однако эта теория в данном случае оказалась неверной. Правильный механизм реакции дал в 1918 немецкий физикохимик лауреат Нобелевской премии Вальтер Нернст. Он предположил, что активными частицами являются атомы водорода и хлора; при этом схема цепной реакции выглядела так. Зарождение цепи происходит при термической диссоциации молекул хлора при высокой температуре или же при поглощении ими квантов света при комнатной температуре: Cl2 ® 2Cl. Далее следуют две быстро повторяющиеся одна за другой стадии продолжения цепи: Cl + H2 ® HCl + H и H + Cl2 ® HCl + Cl. Обрыв цепей происходит, когда активные атомы водорода или хлора реагируют с молекулами примеси, или «прилипают» к стенке сосуда, или реагируют (рекомбинируют) друг с другом, превращаясь в неактивные молекулы H2 и Cl2.
В последующем было показано, что атомы водорода намного активнее атомов хлора; соответственно атомы водорода реагируют намного быстрее и потому их стационарная концентрация значительно ниже. Так, при комнатной температуре стационарная концентрация атомов водорода примерно в 100 раз меньше, чем атомов хлора. В результате вероятность встречи двух атомов водорода или атомов водорода и атомов хлора намного меньше, чем для двух атомов хлора, поэтому практически единственной реакцией обрыва цепей является рекомбинация атомов хлора: Cl + Cl ® Cl2. Если давление в реакционном сосуде очень мало, а его размеры невелики, активные частицы могу достигнуть стенки сосуда еще до того, как прореагируют с молекулами H2 и Cl2; в этих условиях важную роль может приобрести обрыв цепей на стенках реакционного сосуда.
Схема Нернста была подтверждена разными экспериментами. Один из самых остроумных провел английский физикохимик Майкл Поляни. В его опытах струя водорода проходила над слегка подогретым металлическим натрием и уносила с собой некоторое очень небольшое количество его паров. Затем струя попадала в темноте в сосуд с хлором. При температуре опыта чистый водород с хлором не реагировал, но ничтожная примесь паров натрия полностью меняла дело: шла быстрая реакция образования хлороводорода. Здесь роль инициатора цепной реакции вместо света играет натрий: Na + Cl2 ® NaCl + Cl. Подобно тому, как в фотохимической реакции на каждый поглощенный квант света приходится очень много прореагировавших молекул, так и здесь на каждый прореагировавший атом натрия приходится много образовавшихся молекул HCl. Аналогичные результаты Поляни получил для реакции хлора с метаном. В этом случае реакции инициирования и обрыва цепей были такими же, как в реакции хлора с водородом, а реакции продолжения цепи выглядели так: Cl· + CH4 ® HCl + ·CH3 и ·CH3 + Cl2 ® CH4 + Cl·. В этих реакциях также участвуют частицы с неспаренными электронами (обозначены точкой) – свободные радикалы.
Многие реакции с участием свободных радикалов оказались цепными, механизм которых в общих чертах был сходен с механизмом реакции водорода с хлором. По цепному механизму протекают реакции расщепления при высоких температурах (пиролиза) углеводородов, например, этана: C2H6 ® C2H4 + H2; подобные реакции имеют большое значение при промышленной переработке углеводородов нефти. Цепными оказались реакции окисления органических соединений кислородом, реакции присоединения к непредельным соединениям галогенов (хлора и брома), бромоводорода и других соединений, реакции полимеризации, ряд других процессов. Цепные реакции полимеризации интересны тем, что в них стадии продолжения цепи оставляют за собой «реальные цепи» в виде связанных друг с другом остатков мономерных звеньев. В загустевшем и затвердевшем полимере (например, в полистироле или в полиметилметакрилате – «органическом стекле») иногда можно обнаружить концевые свободные радикалы, которым из-за высокой вязкости не удалось прореагировать со свободной молекулой мономера.
Цепи Семенова – Хиншелвуда. В конце 1924 в Ленинградском Физико-техническом институте, в Лаборатории электронной химии, которой заведовал Н.Н.Семенов, начали измерять интенсивность свечения паров фосфора при их окислении кислородом. В первых же опытах молодая выпускница университета Зинаида Вальта и ее непосредственный руководитель, Ю.Б.Харитон натолкнулись на совершенно неожиданное явление. Оказалось, что когда кислорода мало, реакция окисления фосфора вообще не идет. Но стоит давлению кислорода превысить некоторое критическое значение, начиналось интенсивное окисление с испусканием света. До этого теория предполагала, что скорость реакции должна плавно увеличиваться с увеличением концентрации. Здесь же – резкий переход от полного отсутствия реакции к очень быстрому процессу при ничтожном изменении давления. Выяснился еще один, совсем уже странный факт: при давлении ниже критического, т.е. в отсутствие реакции, достаточно было ввести в сосуд аргон, чтобы произошла яркая вспышка. Получалось, что инертный газ аргон, не способный ни к каким химическим реакциям, делал кислород реакционноспособным! Это было уже настоящим чудом...
Позже выяснилось, что кислород может полностью терять свою активность не только при снижении, но и при повышении давления выше некоторого критического значения. Этот второй (верхний) предел давления кислорода существенно зависел от примесей различных веществ. Некоторые из таких примесей делали «пассивный» кислород весьма активным, вызывающим горение фосфора. Такое поведение противоречило всем существовавшим тогда представлениям о механизмах и скоростях химических реакций.
Результаты странных экспериментов, без какой-либо попытки их объяснения, были опубликованы в немецком «Физическом журнале». Последствия были быстры и неутешительны: работа подверглась крайне острой критике со стороны знаменитого Боденштейна, который к тому времени считался главой мировой химической кинетики. Он написал, что все результаты по окислению фосфора являются не открытием, а иллюзией и указал даже на ее причину – неправильную конструкцию установки, в которой проводились опыты. В заключение своей короткой статьи Боденштейн отмечал, что так называемые «предельные» явления неоднократно наблюдались в прошлом для разных реакций, но при проверке каждый раз оказывалось, что все они связаны с различными экспериментальными ошибками.
Возражения были очень серьезны. Но тщательная проверка (уже без Харитона – он был в заграничной командировке и без Вальта – она перешла в другой институт) показала правильность первой публикации. Более того, были получены новые, не менее «еретические» данные. Оказалось, например, что критическое давление кислорода сильно зависит от размеров реакционного сосуда.
Семенов почувствовал, что стоит на пороге открытия. Он понимал, что реакция является цепной, наподобие реакции водорода с хлором. Однако механизм цепной реакции Боденштейна – Нернста, основанный на «принципе домино», никогда не приводил (и не мог приводить) к критическим явлениям. Здесь было что-то иное. Одновременно в этом направлении начал работать в Англии Сирил Хиншелвуд. В обеих лабораториях критические явления были обнаружены в реакциях горения водорода и ряда других веществ. Оказалось, например, что в стеклянных термостойких сосудах при температурах 500–600° С реакция водорода с кислородом не идет вовсе, пока давление не достигнет 3–4 мм рт. ст. Когда давление превышало этот нижний предел, внезапно начиналась быстрая реакция, сопровождающаяся свечением. Но самое поразительное явление заключалось в том, что пламя можно было потушить, просто повысив давление. При температуре же ниже 400° С воспламенение в чистой смеси водорода с кислородом не наблюдалось ни при каких давлениях. Однако достаточно было добавить к смеси инертный газ, как происходила вспышка!
Все эти новые явления были объяснены Семеновым (и независимо Хиншелвудом) в предположении о разветвляющихся цепях. Если в реакции водорода с хлором на каждой стадии продолжения цепи одна активная частица расходуется и одна – появляется (неразветвленная цепь), то в реакции водорода (и других реагентов) с кислородом на одну исчезнувшую активную частицу образуется две или более новых, например, H + O2 ® OH + O O + H2 ® OH + H OH + H2 ® H2O + H
Если сложить эти три последовательные реакции, получим Н + О2 + 2Н2 ® ОН + 2Н, то есть одна активная частица превращается в три. В результате число активных центров стремительно нарастает (цепи разветвляются), и если скорость обрыва цепей недостаточно велика, реакция очень быстро переходит во взрывной режим (при небольшом давлении вместо взрыва наблюдается вспышка). Такие реакции, идущие с увеличение числа активных частиц, назвали разветвленно-цепными. Если учесть, что эти процессы сильно экзотермичны, а для реакции каждой активной частицы с молекулой исходного вещества требуются миллиардные доли секунды, то легко понять, почему разветвленно-цепные реакции при больших концентрациях (давлениях) реагентов вызывают разрушительные взрывы.
Важно отметить, что лавина разветвленно-цепной реакции очень быстро заканчивается: спустя доли секунды после ее начала для продолжения реакции уже не хватает исходных веществ – они почти все превратились в продукты реакции. Здесь можно привести такую аналогию: по «разветвленно-цепному механизму» распространяются различные слухи, если каждый узнавший новость расскажет ее более чем одному человеку. И так же, как слухи и сплетни, быстро распространяются, но и быстро заканчиваются разнообразные «разветвленно-цепные» финансовые и прочие пирамиды (типа знаменитой «Властилины», МММ, «писем счастья» и т.п.), различные «заманчивые» предложения за 100 рублей заработать 100 тысяч и другие аферы, требующие на каждом этапе привлечения новых «клиентов». С первого взгляда все выглядит честно, но в пирамиду быстро вовлекается все большее число участников, и вскоре не хватает «исходных веществ» – некому больше покупать акции, и они стремительно обесцениваются. Подобные финансовые пирамиды еще в 19 в. были в ходу в разных странах; во Франции их называли «снежным комом», у нас – лавиной. Их механизм (и математическое описание) весьма напоминают разветвленно-цепные химические реакции.
Семенов и Хиншелвуд дали такое объяснение изученным процессам. При низких давлениях большинство активных частиц – атомов, свободных радикалов, не успев столкнуться со многими молекулами реагентов и «размножиться», долетают до стенки реакционного сосуда и «погибают» на них – цепи обрываются. Чем меньше диаметр реактора, тем больше у радикалов шансов достичь его стенок. Вот откуда зависимость от размеров сосуда! С повышением концентрации шансов столкнуться с молекулами реагентов для радикалов становится больше, чем шансов достичь стенки – возникает лавина реакций. Это объясняет существование нижнего предела по давлению. Молекулы инертного газа, по меткому выражению Семенова, «путаясь в ногах» у активной частицы, замедляют ее движение к стенке; так объясняется удивительное влияние аргона на величину критического давления.
Когда же достигается верхний предел по давлению, цепи снова обрываются быстрее, чем происходит их разветвление; однако причина обрыва цепей здесь иная – активные радикалы исчезают в результате «взаимного уничтожения» – рекомбинации в объеме сосуда (скорость этой реакции очень быстро увеличивается с ростом давления). Таким образом, все экспериментальные факты получили логичное объяснение в рамках теории разветвленной цепной реакции. В 1956 Н.Н.Семенов и С.Хиншелвуд за эти исследования получили Нобелевскую премию по химии.
Теория разветвленно-цепных реакций имеет большое практическое значение, так как объясняет поведение многих промышленно важных процессов, таких как горение, крекинг нефти, воспламенение горючей смеси в двигателях внутреннего сгорания. Наличие верхнего и нижнего пределов по давлению означает, что смеси кислорода с водородом, метаном, другими горючими газами взрываются лишь при их определенных соотношениях. Например, смеси водорода с воздухом взрываются при содержании водорода от 4 до 75%, а смеси метана с воздухом – при содержании метана от 5 до 15%. Вот почему так опасны утечки газа: если метана в воздухе окажется больше 5%, взрыв может наступить даже от крошечной искры в выключателе при включении или выключении света на кухне.
Особое значение цепные процессы приобрели в связи с работой физиков по получению ядерной энергии. Оказалось, что деление урана, плутония, других расщепляющихся материалов подчиняется тем же закономерностям, что и разветвленно-цепные химические реакции. Так, реакция деления урана вызывается нейтронами, которые расщепляют ядра урана с выделением огромной энергии. Разветвление цепи происходит за счет того, при расщеплении ядра выделяются несколько активных частиц – нейтронов, способных к расщеплению новых ядер.
Реакции с вырожденным разветвлением. При окислении некоторых соединений получаются пероксиды, которые сами способны в определенных условиях распадаться с образованием активных частиц – свободных радикалов. В результате происходит разветвление цепей, хотя и не такое быстрое: ведь чтобы распад пероксидов шел с заметной скоростью, они должны сначала накопиться. Такие процессы назвали вырожденными разветвлениями.
Типичный пример разветвленно-цепной реакции с вырожденным разветвлением – реакция окисления углеводородов. Она начинается с того, что молекула кислорода отрывает от молекулы органического соединения атом водорода: RH + O2 ® R· + HO2·. Гидропероксидный радикал, образовавшийся на стадии инициирования, в результате реакции HO2· + RH ® H2O2 + R· превращается в радикал R· с неспаренным электроном на атоме углерода. Так что радикал HO2· далее в реакции не участвует. У радикала R· несколько возможностей. Во-первых, он может соединяться (рекомбинировать) с другими радикалами, в том числе и себе подобными: R· + R· ® R2. Во-вторых, может отрывать атом водорода от молекулы исходного вещества: R· + R'H ® RH + R'·. Наконец, он может присоединяться по двойной связи молекулы кислорода: R· + O=O ® R–O–O·. Первую реакцию можно не принимать во внимание: вероятность встречи двух активных радикалов очень мала, так как их концентрация ничтожна. Вторая реакция приводит лишь к обмену атомом водорода. А вот в результате третьей реакции образуется пероксидный радикал RO2·, который вместе с радикалом R· ведет цепь. Она состоит из двух повторяющихся стадий цепной реакции окисления: RO2· + RH ® ROOH + R· и R· + O2 ® RO2·.
Видно, что цепь ведут радикалы RO2· и R·, поскольку именно они постоянно рождаются в ходе реакции. Радикалы RO2· менее активные, их концентрация намного выше, поэтому цепь обрывается, когда встречаются два пероксидных радикала. Эта встреча может дать различные продукты, в числе которых пероксиды ROOR (они образуются при рекомбинации пероксидных радикалов), спирты, карбонильные соединения. Если цепи длинные, этих веществ – продуктов рекомбинации – будет немного, а основным продуктом цепной реакции станет гидропероксид ROOH, который иногда удается получить с высоким выходом. Связь О–О в гидропероксидах относительно слабая (более чем вдвое слабее связи С–О в спиртах). При ее разрыве образуются сразу два радикала – RO· и OH·, которые инициируют новые цепи. Получается, что продукт реакции, гидропероксид, одновременно ускоряет ее. Такие реакции называются автокаталитическими.
Возрождение «энергетических цепей». Высказанное Боденштейном и рядом других химиков предположение об «энергетических цепях» не получило экспериментального подтверждения и было на многие десятилетия забыто. Однако в 1963 В.И. Веденеев, А.М. Чайкин и А.Е. Шилов обнаружили, что «энергетические разветвления» возможны в реакциях фторирования ряда соединений. Примером может служить реакция фтора с водородом. В этой реакции на стадии продолжения цепи H + F2 ® HF* + F выделяется так много энергии, что образующаяся «горячая» молекула фтороводорода (она помечена звездочкой) может вызвать разветвление цепи. Происходит это путем передачи избыточной энергии исходным веществам; переносчиком энергии при этом является молекула водорода. Механизм реакции таков: F2 + H2 ® H + HF + F – медленная стадия зарождения цепи F + H2 ® HF + H – две реакции
H + F2 ® HF* + F – продолжения цепи HF* + H2 ® HF + H2* – передача возбуждения
H2* + F2 ® H + HF* + F – разветвление цепи Обрыв цепей происходит на молекулах примесей или на стенках сосуда. Исследование механизма этой реакции позволило создать химический фтор-водородный лазер, в котором источником света (в инфракрасном диапазоне) являются возбужденные молекулы HF.
Комплексные соединения. Строение, номенклатура, применение
Комплексные соединения
Комплексные соединения представляют наиболее распространенную и неоднородную группу химических веществ. К ним относится огромное количество минералов и природных металлоорганических соединений (хлорофилл, гемоглобин, витамин В12 и т.п.), а также множество искусственно синтезированных веществ, которые приобрели важное значение в прикладной химии, химической технологии и почти во всех без исключения областях хозяйства.Учитывая многочисленность комплексных соединений и разнообразие присущих им свойств, не удается сформулировать однозначное определение, которое охватывало бы все разновидности этого класса веществ. Однако инженер в практической деятельности наиболее часто имеет дело с соединениями, для которых справедливой считается такая характеристика.
Комплексными называются соединения, в узлах кристаллических решеток которых размещаются сложные ионы, построенные за счет координации определенных частиц вокруг центрального атома (иона) и способные к самостоятельному существованию при переходе вещества в растворенное или расплавленное состояние. Между комплексными соединениями и некоторыми обычными веществами невозможно провести резкую границу. Более того, одно и то же вещество в зависимости от условий может вести себя поразному. Например, в твердом состоянии роданид состава Pb(CNS)2· 4KCNS содержит в узлах кристаллической решетки сложные ионы [Pb(CNS)6]4-, которые не разрушаются и при его растворении в органических растворителях, способствующих протеканию электролитической диссоциации:
Координационная теория комплексных соединений К концу ХIХ в. был накоплен огромный материал о свойствах комплексных соединений, однако их строение и состав не удавалось объяснить с позиций классической теории валентности.Основатель координационной теории А.Вернер (1893г.) предположил, что в отличие от обычных веществ в комплексных соединениях элементы проявляют, кроме главной валентности, еще и дополни-тельную, побочную. Благодаря действию сил именно побочной валентности происходит процесс комплексообразования. Но в самом комплексе различие между главной и побочной валентностями исчезает, все связи становятся равноценными, их называют координационными.
Вернер подчеркнул, что действие побочной валентности вызывает укрепление связей между атомами. Например, соединение PbCl4 чрезвычайно неустойчиво, а комплексное соединение K2[PbCl6], образование за счет побочной валентности свинца(+2), -- довольно прочное. Таким образом, в комплексных соединениях наблюдается стабилизация как высших, так и низших валентных форм элемента.
В современном виде координационная теория А.Вернера, которая постоянно углубляется и дополняется, сводится к нескольким основным положениям.
1. В молекуле комплексного соединения атом (ион), который за счет главной и побочной валентностей координирует вокруг себя определенное количество нейтральных молекул или противоположно заряженных ионов, называется центральным атомом, или комплексообразователем.
Наиболее часто в роли комплексообразователя выступают положительно заряженные ионы (реже -- атомы) d- и р-металлов, иногда -- щелочноземельных и даже щелочных металлов, а также некоторых неметаллов ( В, Si, P, As).
2. С центральным атомом непосредственно соединяются молекулы или ионы, которые называются координированными группами, аддендами, или лигандами.
3. Комплексообразователь совместно с лигандами составляет внутреннюю сферу комплексного соединения, или просто комплекс, который при написании координационной формулы берется в квадратные скобки, чтобы подчеркнуть его монолитность: К3[Fe(CN)6], Na[BF4].
Заряд внутренней сферы определяется алгебраической суммой степени окисления комплексообразователя и совместных зарядов всех лигандов:
4. Если степень окисления комплексообразователя по абсолютной величине не равна сумме зарядов всех лигандов, то комплексное соединение содержит внешнюю сферу, которую записывают вне квадратных скобок: [Ag(CN)2]Cl, K[Ag(NH3)2]. Однако существуют и нейтральные комплексы, лишенные внешней сферы, их иногда называют комплексными неэлектролитами:
5. Общее количество координационных валентностей, с помощью которых комплексообразователь во внутренней сфере связан с лигандами, называется координационным числом (к.ч.).
Известны координационные числа от 1 до 9 и 12. Наиболее распространенными являются комплексные соединения с координационными валентностями 2, 4 и 6. Довольно часто координационное число бывает вдвое большим, чем степень окисления комплексообразователя, например:
однако это нестрогая зависимость.
В общем случае координационное число может иметь переменную величину не только для разных комплексообразователей, но и для одного и того же, поскольку на количество координированных лигандов влияют определенные факторы:
1. Заряд лиганда. Например, координационное число никеля (+2) с нейтральными лигандами равно 6, а с отрицательно заряженными анионами CN-, между которыми действуют значительные силы отталкивания, -- только 4:
2. Размеры лигандов. Комплексы, содержащие небольшие по размерам ионы, характеризуются большими величинами координационных чисел, например, для фторсодержащих алюминатных комплексов координационное число больше, чем для хлор- и бромсодержащих, так как радиус фторид-иона намного меньше, чем радиусы ионов Cl- и Br-.
Свойства растворителя, в котором происходит образование комплексного соединения. Так, в полярном растворителе -- воде -- легче возникают комплексы [Co(CNS)1]+ и [Co(CNS)2], а в малополярном -- ацетоне -- при одних и тех же условиях -- [Co(CNS)3]- и [Co(CNS)4]2-
· Концентрация реагирующих компонентов. При возрастании концентрации появляется тенденция к образованию более сложных комплексов. Например, в разбавленных растворах удается идентифицировать лишь комплекс [Fe(CNS)1]2+, в то время, как с увеличением концентраций реагирующих веществ получаются более сложные комплексы: от [Fe(CNS)3] вплоть до [Fe(CNS)6]3-
Энергетические эффекты в химических реакциях. Внутренняя энергия. Энтальпия. Стандартная энтальпия образования химических соединений
Энергетические эффекты химических реакций изучает термохимия. Данные об энергетических эффектах используются для выяснения направленности химических процессов, для расчета энергетических балансов технологических процессов и т.д. С их помощью можно рассчитать температуру горения различных веществ и материалов, температуру пожаров и т.п.
Состояние системы (вещества или совокупности рассматриваемых веществ) описывают с помощью ряда параметров состояния – t, p, m. Для характеристики состояния системы и происходящих в ней изменений важно знать также изменение таких свойств системы, как ее внутренняя энергия U, энтальпия Н, энтропия S, энергия Гиббса (изобарно-изотермический потенциал) G. По изменению этих свойств системы можно судить, в частности, об энергетике процессов.
Химические реакции обычно протекают при постоянном объеме V = const, DV = 0 (например, в автоклаве) или при постоянном давлении p = const (например, в открытой колбе), т.е. является соответственно изохорными или изобарными процессами.
Энергетический эффект химического процесса возникает за счет изменения в системе внутренней энергии U или энтальпии H. Внутренней энергией системы называют энергию всех видов движения и взаимодействия тел или частиц, составляющих систему (кинетическая энергия межмолекулярного взаимодействия, вращательная энергия, колебательное движение атомов и групп в молекуле, энергия взаимодействия электронов между собой и с ядрами).
Обычно химические реакции сопровождаются тепловыми эффектами. Тепловым эффектом называется суммарное количество энергии, выделенной или поглощенной системой в результате реакции, проводимой при постоянной температуре. Раздел химии, который изучает тепловые эффекты химических реакций и фазовых превращений, называется термохимией.
Согласно первому закону термодинамики (уравнение 4.6) количество выделенной или поглощенной системой теплоты Q определяется равенством:
Q = DU + W.
Подставив выражение (4.5) в (4.6), получим равенство:
Q = DU + p·DV, (4.7).
определяющее тепловой эффект химической реакции. Из равенства (4.7) следует, что тепловой эффект реакции зависит от того, в каких условиях она протекает. В изохорном процессе V = const, DV = 0, следовательно, тепловой эффект реакции QV равен изменению внутренней энергии системы:QV = U2 – U1 = DU, т.к. W = 0 (4.8).
В изобарном процессе p = const, следовательно, тепловой эффект реакции Q равен:
QP = DU + p·DV = (U2 – U1) + p·(V2 – V1) = (U2 + p·V1) - (U1 + p·V1).
Обозначим:U + p·DV = H (4.9).
Величина H называется энтальпией или теплосодержанием системы. Поэтому тепловой эффект химической реакции при изобарном процессе равен изменению энтальпии системы:
QP = H2 – H1 = DH (4.10).
или
QP = DU + p·DV = DH (4.10а).
Энтальпия, также как и внутренняя энергия, является термодинамической функцией состояния системы.
Для реакций, в которых участвуют только твердые и жидкие вещества, член p·DV в уравнении (4.10а) пренебрежимо мал или равен нулю. Для подобных реакций выполняется соотношение DH » DU. Для газофазных реакций, протекающих с участием газообразных веществ, изменение объема значительно. Если DV > 0, т.е. происходит расширение, то DH > DU; если DV < 0, т.е. происходит сжатие, то DH < DU. Произведение p·DV для таких реакций можно рассчитать из уравнения идеального газа:p·DV = n·R·T или
p·DV = Dn·R·T, где Dn - изменение числа моль газа, определяемое из уравнения реакции; например,
(NH4)2Cr2O7 = Cr2O3 + N2 + 4H2O, Dn = 5.
Химические реакции, протекающие с выделением теплоты, называются экзотермическими. При этом в изохорном процессе внутренняя энергия системы уменьшается, т.е. DU < 0 (т.к. U2 < U1), а в изобарном процессе - энтальпия уменьшается, т.е. DH < 0 (т.к. H2 < H1) (рис.4.2).
Химические реакции, протекающие с поглощением теплоты, называются эндотермическими. При этом в изохорном процессе DU > 0, в изобарном процессе - DH > 0. Уменьшение энтальпии в экзотермических процессах означает, что суммарная энергия, содержащаяся в продуктах реакции в виде энергии химических связей, межмолекулярных взаимодействий, молекулярных колебаний и т.д. меньше суммарной энергии исходных веществ (реагентов). И наоборот, увеличение энтальпии в эндотермических процессах означает, что суммарная энергия, содержащаяся в продуктах реакции больше суммарной энергии исходных веществ.
Изменение энтальпии при стандартном состоянии веществ, участвующих в реакции или при фазовом превращении, обозначается DH°(T) и DH°(298 K), если температура системы T или 298,15 K.
Тепловые эффекты химических реакций зависят не только от условий (температура, давление, объем), в которых они протекают, но и от количества веществ, участвующих в реакции, и их физического состояния. Поэтому для того, чтобы можно было сравнивать энергетические эффекты различных процессов, их характеризуют изменением энтальпии при стандартных условиях, соответствующим конкретному уравнению химической реакции. Уравнения химических реакций, в которых указаны их тепловые эффекты и агрегатные состояния (г-газовое, ж-жидкое, к-кристаллическое, т-твердое) или аллотропные модификации (например, a-сера, b-сера) веществ, называются термохимическими уравнениями реакций. Например:
2H2(г) + O2(г) = 2H2O(ж), DH°(298 K) = -571,6 кДж 2H2(г) + O2(г) = 2H2O(г), DH°(298 K) = -483,6 кДж
ТЕРМОХИМИЧЕСКИЕ ЗАКОНЫ. ТЕРМОХИМИЧЕСКИЕ РАСЧЕТЫ
Тепловые эффекты химических реакций можно определить экспериментально или расчетным путем. Измерение тепловых эффектов называется калориметрией. В основе термохимических расчетов лежит закон, сформулированный русским ученым Г.И. Гессом (1840 г.):
Тепловой эффект химической реакции не зависит от пути ее протекания, а зависит от природы и физического состояния исходных веществ и продуктов реакции.Это означает, что если какую-либо реакцию представить в виде нескольких последовательных стадий, то тепловой эффект данной реакции будет равен сумме тепловых эффектов каждой стадии.
Например, тепловой эффект реакции горения метана равен DH° = -890,2 кДж
(1) CH4 + 2O2 = CO2 + 2H2O ; DH°1 = -890,2 кДжПусть это превращение представляет собой “Путь А”, проходящий через стадию (1). Можно представить протекание данной реакции через “Путь В”, проходящий через ряд промежуточных стадий (2), (3), (4) и (5), где стадия (5) = (3) + (4). Тепловые эффекты каждой из этих стадий равны соответственно:
(2) CH4(г) = C(графит) + 2H2(г) ; DH°2 = +74,9 кДж
(3) C(графит) + O2(г) = CO2(г) ; DH°3 = -393,5 кДж
(4) 2H2(г) + O2(г) = 2H2O(ж) ; DH°4 = -571,6 кДж
Согласно закону Гесса сумма тепловых эффектов на каждой стадии “Пути В” будет равна тепловому эффекту реакции горения метана на “Пути А”:
DH°1 = DH°2 + DH°5= DH°2 + DH°3 + DH°4 -890,2 = 74,9 - 393,5 - 571,6 (кДж)
Экспериментально было установлено (закон Ломоносова - Лавуазье - Лапласа), что тепловые эффекты прямой и обратной реакций численно равны, но противоположны по знаку.
Так, если прямая реакция экзотермическая, то обратная - эндотермическая:
Из закона Гесса вытекают два важных в практическом отношении следствия.
Первое следствие из закона Гесса: тепловой эффект реакции равен сумме энтальпий (теплот) образования продуктов реакции за вычетом суммы энтальпий (теплот) образования исходных веществ с учетом стехиометрических коэффициентов в уравнении реакции.
Так для реакции, протекающей по уравнению:
aA + bB = pP + qQ,
тепловой эффект рассчитывается по формуле:
DH = [pDfH(P) + qDfH(Q)] - [aDfH(A) + bDfH(B)] (4.11).
Энтальпия (теплота) образования - это тепловой эффект реакции образования 1 моль сложного вещества из простых веществ: DfH [Дж/моль; кДж/моль]. Обычно в расчетах используют стандартные энтальпии образования. Стандартная энтальпия образования DfH°(298 K) это тепловой эффект образования 1 моль сложного вещества из простых веществ при стандартных условиях (T = 298,15 K и p = 101,3 кПа). Стандартные энтальпии образования простых веществ, устойчивых в стандартных условиях (газообразный O2, кристаллический I2 и т.д.) принимают равными нулю. Например, окисление водорода можно представить тремя уравнениями:
(6) 2H2(г) + O2(г) = 2H2O(ж) ; DH°(298K)6 = -571,6 кДж
(7) H2(г) + 1/2O2(г) = H2O(ж) ; DH°(298K)7 = -285,8 кДж
(8) H2(г) + 1/2O2(г) = H2O(г) ; DH°(298K)8= -241,8 кДж
Каждому уравнению соответствует определенное значение теплового эффекта. И только тепловой эффект реакции, описываемой уравнением (7), будет равен стандартной теплоте образования воды DH°(298 K)7 = DfH°(298 K, H2O(ж)). Согласно этому уравнению в реакции образуется 1 моль воды, стандартным состоянием которой при 298 K является жидкое.Для многих веществ стандартные теплоты образования известны и сведены в справочные таблицы.Теплота образования является мерой термодинамической устойчивости (прочности) сложного вещества относительно простых веществ, из которых оно образовано. Можно утверждать, что чем более отрицательное значение имеет стандартная энтальпия образования вещества, тем оно устойчивее. Согласно закону Ломоносова - Лавуазье - Лапласа теплота (энтальпия) образования сложного вещества равна по величине, но противоположна по знаку теплоте (энтальпии) разложения вещества (DdH°(Т)):DfH°(298 K) = |-DdH°(298 K)| Второе следствие из закона Гесса: тепловой эффект реакции равен сумме энтальпий (теплот) сгорания исходных веществ за вычетом суммы энтальпий (теплот) сгорания продуктов реакции с учетом стехиометрических коэффициентов в уравнении реакции.
Так для реакции, протекающей по уравнению: aA + bB = pP + qQ тепловой эффект можно рассчитать по уравнению:DH = [aDсH(A) + bDсH(B)] - [pDсH(P) + qDсH(Q)] Энтальпия (теплота) сгорания - это тепловой эффект сгорания 1 моль горючего вещества до продуктов предельного окисления (до образования высших оксидов): DcH [Дж/моль, кДж/моль].Стандартная энтальпия сгорания DcH°(298 K) - это тепловой эффект реакции сгорания в кислороде 1 моль данного вещества при стандартных условиях. Например, тепловой эффект реакции сгорания 1 моль метана в стандартных условиях равен стандартной теплоте сгорания метана:CH4(г) + 2O2(г) = CO2(г) + 2H2O(г) ; DH°(298 K) = -890,2 кДж, т.е. DH°(298 K) = DcH(298 K, CH4(г)) = -890,2 кДж/моль.Энтальпии сгорания кислорода и высших оксидов равны нулю. Следует четко разграничивать два различных понятия - энтальпия образования и энтальпия сгорания вещества, хотя численные значения этих величин в некоторых случаях совпадают (табл. 4.1). Например, стандартные энтальпии сгорания водорода и углерода равны стандартным энтальпиям образования CO2 и H2O.
Закон Гесса распространяется не только на химические реакции, но и на различные физико-химические процессы, сопровождающиеся энергетическими эффектами: растворение, сольватацию (гидратацию), фазовые превращения (плавление, испарение, возгонка, затвердевание (кристаллизация), конденсация, сублимация)Закон Гесса и его следствия справедливы и используются также для расчета энергии химической связи, энергии кристаллической решетки, энергии межмолекулярного взаимодействия, энтальпии растворения и сольватации (гидратации) и т.д. Закон Гесса справедлив для тех взаимодействий, которые протекают при постоянном объеме или при постоянном давлении, а единственным видом совершаемой работы является работа против сил внешнего давления. Тепловые эффекты реакций, в результате которых совершается другая работа, например, электрическая работа, не могут быть вычислены по закону Гесса, так как их теплоты являются функциями пути.
В каждом веществе запасено определенное количество энергии. С этим свойством веществ мы сталкиваемся уже за завтраком, обедом или ужином, так как продукты питания позволяют нашему организму использовать энергию самых разнообразных химических соединений, содержащихся в пище. В организме эта энергия преобразуется в движение, работу, идет на поддержание постоянной (и довольно высокой!) температуры тела.
Энергия химических соединений сосредоточена главным образом в химических связях. Чтобы разрушить связь между двумя атомами, требуется ЗАТРАТИТЬ ЭНЕРГИЮ. Когда химическая связь образуется, энергия ВЫДЕЛЯЕТСЯ.
Вспомним, что атомы не соединялись бы между собой, если бы это не вело к "выигрышу" (то есть высвобождению) энергии. Этот выигрыш может быть большим или малым, но он обязательно есть при образовании молекул из атомов.
Любая химическая реакция заключается в разрыве одних химических связей и образовании других.
Когда в результате химической реакции при образовании новых связей выделяется энергии БОЛЬШЕ, чем потребовалось для разрушения "старых" связей в исходных веществах, то избыток энергии высвобождается в виде тепла. Примером могут служить реакции горения. Например, природный газ (метан CH4) сгорает в кислороде воздуха с выделением большого количества теплоты (рис. 1-1а). Такие реакции называются ЭКЗОТЕРМИЧЕСКИМИ от латинского "экзо" - наружу (имея в виду выделяющуюся энергию).
В других случаях на разрушение связей в исходных веществах требуется энергии больше, чем может выделиться при образовании новых связей. Такие реакции происходят только при подводе энергии извне и называются ЭНДОТЕРМИЧЕСКИМИ (от латинского "эндо" - внутрь). Примером является образование оксида углерода (II) CO и водорода H2 из угля и воды, которое происходит только при нагревании
Термохимия. Закон Гесса. Следствия из закона Гесса. Теплотворная способность топлив
Раздел химии, посвящённый изучению тепловых эффектов реакций, получил название термохимия.
Термохимия базируется на 2-х основных законах: Если при образовании какого-либо соединения выделяется (или поглощается) некоторое количество теплоты, то при разложении этого соединения в тех же самых условиях такое же количество теплоты поглощается (или выделяется). Результаты термохимических изменений - тепловые эффекты реакций, относят к одному молю образующегося вещества, а количество теплоты, которое выделится при образовании одного моля соединения из простых веществ, называют теплотой образования данного соединения. Если элемент может существовать в виде нескольких простых веществ или алотропных форм, то при расчете теплоты образования этот элемент берут в виде того вещества, которое при данных условиях наиболее устойчиво. Теплообразование наиболее устойчивых при данных условиях простых веществ принимают равным нулю. Теплотообразование менее устойчивых простых веществ равны теплотам их образования из наиболее устойчивых. Химические уравнения, в которых указано количеств выделяющейся, или поглощаемой теплоты, а также агрегатные состояния реагирующих веществ, называют термохимическими уравнениями. Величина теплового эффекта зависит от природы реагирующих веществ, продуктов реакции, их агрегатного состояния и температуры. Для удобства сравнения различных реакций по величинам их тепловых эффектов последние, как правило, указываются для температур исходных веществ, равной 25 ˚С или 298 К, называемой стандартной температурой.
Важнейшей характеристикой веществ, применяемых в качестве топлива служит теплота их сгорания, которая характеризует количество теплоты, выделяющейся при сгорании 1 моля вещества.
Закон Гесса: тепловые эффекты реакций зависят только от начального и конечного состояний веществ и не зависят от промежуточных стадий процесса. Следствие из закона Гесса: тепловой эффект химической реакции равен суме теплот образования получившихся веществ за вычетом суммы теплот образования исходных веществ. Полу-суммирование осуществляется с учётом числа молей, участвующих в реакции веществ в соответствии с её уравнением. Теплотворная способность топлива Качество топлива характеризуется его теплотворной способностью Q и измеряется количеством тепла, выде¬ляющегося при полном сгорании 1 кг твердого или жидкого или I нормального кубического метра (1 нм3)1 газообразного топ¬лива. Для твердого и жидкого топлива эта величина имеет размерность ккал1кг, для газообразного — ккал/нм3.Теплотворная способность рабочей массы топлива обозна¬чается: <?р ккал/кг — для твердого и жидкого топлива и Qp ккал1нм3 — для газообразного.Принято считать, что если теплотворная способность топли¬ва учитывает, кроме тепла, выделившегося при его сгорании, также и тепло, полученное при преобразовании продуктов го¬рения в воду при 100°С, то такая теплотворность называется высшей (QP).
Если при определении теплотворной способности топлива было учтено тепло, расходованное на испарение влаги, такая теплотворность называется низшей (Qp). При практических расчетах пользуются последней.
Теплотворная способность твердого и жидкого топлива определяется либо опытным путем, либо расчетным по формуле Д. И. Менделеева:
Qp = 81Ср + 300НР — 26(0? — SP) ККАЛ/КГ;
Qp = 81Ср + 300НР — 26(Ор — Sp) — 6(9Нр +WP) ККАЛ/КГ.
Теплотворная способность газообразного топлива определя¬ется по формулам, полученным методом суммирования произ¬ведений тепловых эффектов горения газов на их процентное содержание:
QB = 30.5СО + 30,5Н2 + 95,ЗСН4 + 152,5С2Н4 + 4- 60,0H2S ККАЛ/НМ3;
Q„ = 30.5СО + 25,8Н2 + 85,9СН4 + 143,0С2Н4 + 4- 55,2H2S ККАЛ/НМ3.
Для сравнения тепловой ценности различных видов топлива применяется общая единица измерения—так называемое услов¬нее топливо, теплотворная способность которого 7000 ккал/кг. Для перевода любого топлива в условное необходимо действи¬тельную теплотворную способность разделить на 7000 ккал/кг. Полученное отношение называется калорийным эк¬вивалентом. Естественно, что чем больше калорийный эк¬вивалент топлива, тем оно ценнее.Теплотворная способность и калорийные эквиваленты не¬которых наиболее распространенных видов топлива приведены в табл. у. Сжигание топлива и использование тепла отходящих газовТеплотворная способность и калорийные эквиваленты некоторых видов топлива
Горение топлива начинается только в том случав,- когда тем¬пература в топливосжигающем устройстве доведена до темпе¬ратуры воспламенения. Последняя величина переменная и зависит от вида топлива и условий, в которых совершается процесс горения.Горение топлива считается полным, если в продуктах горе¬ния произошло окисление горючих составляющих до ССЬ, ШО, SO2 и -при этом отсутствует свободный кислород.Зная состав топлива, можно подсчитать по формулам, при¬веденным в специальной литературе, теоретическое количество воздуха, необходимое для полного его сжигания. Так как прак¬тически нельзя достичь равномерного смешения воздуха с го¬рючими составляющими топлива, то для обеспечения полного его сгорания необходимо подвести большее количество возудха по сравнению с теоретически необходимым.Отношение количества практически подведенного воздуха 1пр к количеству теоретически необходимому LTeop называется коэффициентом" избытка воздуха:
Величина коэффициента избытка воздуха колеблется в пре¬делах от 1,03 до 1,7. Нижний предел относится к газообразно¬му и жидкому топливу, верхний — к твердому топливу.
Следует помнить, что увеличение избытка воздуха вызывает дополнительный расход тепла для его нагрева, а также увели¬чение количества отходящих продуктов горения, которые уносят большое количество тепла в атмосферу и тем самым снижают температуру нагревательной среды печи. Поэтому необходимо всегда стремиться к полному сжиганию топлива при минималь¬ном коэффициенте избытка воздуха.
Критерием полноты горения служит цвет пламени. При не¬достатке воздуха пламя длинное, красного цвета с черными прожилками. Большой недостаток воздуха вызывает неполное сгорание топлива и появление черного дыма.При избытке воздуха получается короткое, острое (колючее) пламя с ярко светящимися язычками. Такое пламя нагревает маталл неравномерно, вызывает местный его перегрев и даже оплавление. При этом увеличивается расход топлива, угар ме¬талла и вместе с тем снижается температура в печи.
Пламя молочно-белого цвета, без ярких языков в топливо-сжигающем устройстве является признаком оптимального количества воздуха.
Энтропия и ее изменение при химических процессах. Вычисление изменения энтропии.
Энтропия (от греч. entropнa — поворот, превращение), понятие, впервые введенное в термодинамике для определения меры необратимого рассеяния энергии. Э. широко применяется и в других областях науки: в статистической физике как мера вероятности осуществления какого-либо макроскопического состояния; в теории информации как мера неопределенности какого-либо опыта (испытания), который может иметь разные исходы. Эти трактовки Э. имеют глубокую внутреннюю связь. Например, на основе представлений об информационной Э. можно вывести все важнейшие положения статистической физики.В термодинамике понятие «Э.» было введено Р. Клаузиусом (1865), который показал, что процесс превращения теплоты в работу следует общей физической закономерности — второму началу термодинамики. Его можно сформулировать строго математически, если ввести особую функцию состояния — Э.Так, для термодинамической системы, совершающей квазистатически (бесконечно медленно) циклический процесс, в котором система последовательно получает малые количества теплоты dQ при соответствующих значениях абсолютной температуры Т, интеграл от «приведенного» количества теплоты dQ/ Т по всему циклу равен нулю(, т. н. равенство Клаузиуса).
Это равенство, эквивалентное второму началу термодинамики для равновесных процессов, Клаузиус получил, рассматривая произвольный циклический процесс как сумму очень большого, в пределе бесконечного, числа элементарных обратимых Карно циклов. Математически равенство Клаузиуса необходимо и достаточно для того, чтобы выражение
dS = dQ/T (1)
представляло собой полный дифференциал функции состояния S, названное «Э.» (дифференциальное определение Э.). Разность Э. системы в двух произвольных состояниях А и В (заданных, например, значениями температур и объемов) равна (2)
(интегральное определение Э.). Интегрирование здесь ведется вдоль пути любого квазистатического процесса, связывающего состояния А и В, при этом, согласно равенству Клаузиуса, приращение Э. DS = SB — SA не зависит от пути интегрирования.Т. о., из второго начала термодинамики следует, что существует однозначная функция состояния S, которая при квазистатических адиабатных процессах (dQ = 0) остаётся постоянной. Процессы, в которых Э. остаётся постоянной, называются изоэнтропийными. Примером может служить процесс, широко используемый для получения низких температур, — адиабатное размагничивание (см. Магнитное охлаждение). При изотермических процессах изменение Э. равно отношению сообщенной системе теплоты к абсолютной температуре. Например, изменение Э. при испарении жидкости равно отношению теплоты испарения к температуре испарения при условии равновесия жидкости с её насыщенным паром. Согласно первому началу термодинамики (закону сохранения энергии), dQ = dU+pdV, т. е. сообщаемое системе количество теплоты равно сумме приращения внутренней энергии dU и совершаемой системой работы pdV, где р — давление, V — объём системы. С учётом первого начала термодинамики дифференциальное определение Э. принимает вид, (3) откуда следует, что при выборе в качестве независимых переменных внутренней энергии U и объёма V частные производные Э. связаны с абсолютной температурой и давлением соотношениями: Эти выражения представляют собой уравнения состояния системы (первое — калорическое, второе — термическое). Уравнение (4) лежит в основе определения абсолютной температуры (см. также Температура, Температурные шкалы). Формула (2) определяет Э. лишь с точностью до аддитивной постоянной (т. е. оставляет начало отсчёта Э. произвольным). Абсолютное значение Э. позволяет установить третье начало термодинамики, или Нернста теорему: при стремлении абсолютной температуры к нулю разность DS для любого вещества стремится к нулю независимо от внешних параметров. Поэтому: Э. всех веществ при абсолютном нуле температуры можно принять равной нулю (эту формулировку теоремы Нернста предложил в 1911 М. Планк). Основываясь на ней, за начальную точку отсчёта Э. принимают So = 0 при Т = 0. Важность понятия Э. для анализа необратимых (неравновесных) процессов: также была показана впервые Клаузиусом. Для необратимых процессов интеграл от приведённой теплоты dQ / Т по замкнутому пути всегда отрицателен (, т. н. неравенство Клаузиуса). Это неравенство — следствие теоремы Карно: кпд частично или полностью необратимого циклического процесса всегда меньше, чем кпд обратимого цикла. Из неравенства Клаузиуса вытекает, что поэтому Э. адиабатически изолированной системы при необратимых процессах может только возрастать. Т. о., Э. определяет характер процессов в адиабатической системе: возможны только такие процессы, при которых Э. либо остаётся неизменной (обратимые процессы), либо возрастает (необратимые процессы). При этом не обязательно, чтобы возрастала Э. каждого из тел, участвующего в процессе. Увеличивается общая: сумма Э. тел, в которых процесс вызвал изменения. Термодинамическому равновесию адиабатической системы соответствует состояние с максимумом Э. Энтропия может иметь не один, а несколько максимумов, при этом система будет иметь несколько состояний равновесия. Равновесие, которому соответствует наибольший максимум Э., называется абсолютно устойчивым (стабильным). Из условия максимальности Э. адиабатические системы в состоянии равновесия вытекает важное следствие: температура всех частей системы в состоянии равновесия одинакова. Понятие «Э.» применимо и к термодинамически неравновесным состояниям, если отклонения от термодинамического равновесия невелики и можно ввести представление о локальном термодинамическом равновесии в малых, но ещё макроскопических объёмах. Такие состояния можно охарактеризовать термодинамическими параметрами (температурой, давлением и т. д.), слабо зависящими от пространственных координат и времени, а Э. термодинамически неравновесного состояния определить как Э. равновесного состояния, характеризующегося теми же значениями параметров. В целом Э. неравновесной системы равна сумме Э. её частей, находящихся в локальном равновесии. Термодинамика неравновесных процессов позволяет более детально, чем классическая термодинамика, исследовать процесс возрастания Э. и вычислить количество Э., образующейся в единице объёма в единицу времени вследствие отклонения системы от термодинамического равновесия — производство энтропии. Производство Э. всегда положительно и математически выражается квадратичной формой от градиентов термодинамических параметров (температуры, гидродинамической скорости или концентраций компонентов смеси) с коэффициентами, называемыми кинетическими (см. Онсагера теорема).Статистическая физика связывает Э. с вероятностью осуществления данного макроскопического состояния системы. Э. определяется через логарифм статистического веса W данного равновесного состояния S= k ln W (E, N), (7) где k — Больцмана постоянная, W (E, N) — число квантовомеханических уровней в узком интервале энергии DЕ вблизи значения энергии Е системы из N частиц. Впервые связь Э. с вероятностью состояния системы была установлена Л. Больцманом в 1872: возрастание Э. системы обусловлено её переходом из менее вероятного состояния в более вероятное. Иными словами, эволюция замкнутой системы осуществляется в направлении наиболее вероятного распределения энергии по отдельным подсистемам. В отличие от термодинамики статистическая физика рассматривает особый класс процессов — флуктуации, при которых система переходит из более вероятного состояния в менее вероятное, и её Э. уменьшается. Наличие флуктуаций показывает, что закон возрастания Э. выполняется только в среднем для достаточно большого промежутка времеЭ. в статистической физике тесно связана с информационной Э., которая служит мерой неопределённости сообщений данного источника (сообщения описываются множеством величин х1, x2,..., xn, которые могут быть, например, словами какого-либо языка, и соответствующих вероятностей p1, p2,..., pn появления величин x1, x2,..., xn в сообщении). Для определённого (дискретного) статистического распределения вероятностей рк информационной Э. называют величину при условии Значение Ни равно нулю, если какое-либо из pk равно 1, а остальные — нулю, т. е. неопределённость в информации отсутствует. Э. принимает наибольшее значение, когда pk равны между собой и неопределённость в информации максимальна. Информационная Э., как и термодинамическая, обладает свойством аддитивности (Э. нескольких сообщений равна сумме Э. отдельных сообщений). К. Э. Шеннон показал, что Э. источника информации определяет критическое значение скорости «помехоустойчивой» передачи информации по конкретному каналу связи (см. Шеннона теорема). Из вероятностной трактовки информационной Э. могут быть выведены основные распределения статистической физики: каноническое Гиббса распределение, которое соответствует максимальному значению информационной Э. при заданной средней энергии, и большое каноническое распределение Гиббса — при заданных средней энергии и числа частиц в системе. Понятие Э., как показал впервые Э. Шрёдингер (1944), существенно и для понимания явлений жизни. Живой организм с точки зрения протекающих в нём физико-химических процессов можно рассматривать как сложную открытую систему, находящуюся в неравновесном, но стационарном состоянии. Для организмов характерна сбалансированность процессов, ведущих к росту Э., и процессов обмена, уменьшающих её. Однако жизнь не сводится к простой совокупности физико-химических процессов, ей свойственны сложные процессы саморегулирования. Поэтому с помощью понятия Э. нельзя охарактеризовать жизнедеятельность организмов в целом. Д.Н. Зубарев. Э., характеризуя вероятность осуществления данного состояния системы, согласно (7) является мерой его неупорядоченности. Изменение Э. DS обусловлено как изменением р, V и Т, так и процессами, протекающими при р, Т = const и связанными с превращением веществ, включая изменение их агрегатного состояния, растворение и химическое взаимодействие. Изотермическое сжатие вещества приводит к уменьшению, а изотермическое расширение и нагревание — к увеличению его Э., что соответствует уравнениям, вытекающим из первого и второго начал термодинамики (см. Термодинамика): Формулу (11) применяют для практического определения абсолютного значения Э. при температуре Т, используя постулат Планка и значения теплоёмкости С, теплот и температур фазовых переходов в интервале от 0 до Т К. В соответствии с (1) Э. измеряется в кал/(моль К) (энтропийная единица — э. е.) и дж/(моль·К). При расчётах обычно применяют значения Э. в стандартном состоянии, чаще всего при 298,15 К (25 °С), т. е. S0298; таковы приводимые ниже в статье значения Э.
Э. увеличивается при переходе вещества в состояние с большей энергией. D S сублимации > DS парообразования >> DS плавления >DS полиморфного превращения. Например, Э. воды в кристаллическом состоянии равна 11,5, в жидком — 16,75, в газообразном — 45,11 э. е.
Чем выше твёрдость вещества, тем меньше его Э.; так, Э. алмаза (0,57 э. е.) вдвое меньше Э. графита (1,37 э. е.). Карбиды, бориды и другие очень твёрдые вещества характеризуются небольшой Э. Э. аморфного тела несколько больше Э. кристаллического. Возрастание степени дисперсности системы также приводит к некоторому увеличению её Э. Э. возрастает по мере усложнения молекулы вещества; так, для газов N2О, N2O3 и N2O5 Э. составляет соответственно 52,6; 73,4 и 85,0 э. е. При одной и той же молекулярной массе Э. разветвленных углеводородов меньше Э. неразветвлённых; Э. циклоалкана (циклана) меньше Э. соответствующего ему алкена. Э. простых веществ и соединений (например, хлоридов ACIn), а также её изменения при плавлении и парообразовании являются периодическими функциями порядкового номера соответствующего элемента. Периодичность изменения Э. для сходных химических реакций типа 1/n Акрист + 1/2Сl2газ = 1/n ACln крист практически не проявляется. В совокупности веществ-аналогов, например АСl4газ (А — С, Si, Ge, Sn, Pb) Э. изменяется закономерно. Сходство веществ (N2 и СО; CdCl2 и ZnCl2; Ag2Se и Ag2Te; ВаСОз и BaSiO3; PbWO4 и РЬМоО4) проявляется в близости их Э. Выявление закономерности изменения Э. в рядах подобных веществ, обусловленного различиями в их строении и составе, позволило разработать методы приближённого расчёта Э. Знак изменения Э. при химической реакции DS х. р. определяется знаком изменения объёма системы DV х. р.; однако возможны процессы (изомеризация, циклизация), в которых DS х. р. № 0, хотя DV х. р. » 0. В соответствии с уравнением DG = DН — ТDS (G — гиббсова энергия, Н — энтальпия) знак и абсолютное значение DS х. р. важны для суждения о влиянии температуры на равновесие химическое. Возможны самопроизвольные экзотермические. процессы (DG < 0, DH < 0), протекающие с уменьшением Э. (DS < 0). Такие процессы распространены, в частности, при растворении (например, комплексообразование), что свидетельствует о важности химических взаимодействий между участвующими в них веществами.
Энергия Гиббса и ее изменение при химических процессах. Условия самопроизвольного протекания химических реакций
Организм совершает работу, затрачивая внутреннюю энергию, запасенную в виде энергии химического взаимодействия атомов составляющих его веществ. Математическое выражение –ДE = –Q – W первого начала термодинамики определяет точное соотношение между расходом внутренней энергии системы ДЕ, работой W, совершаемой системой, и энергией Q, которая теряется в виде теплоты. Однако из первого начала термодинамики нельзя определить часть расходуемой внутренней энергии, которая может быть преобразована в работу. Теоретические оценки затрат осуществляются на основе второго начала термодинамики. Этот закон накладывает строгие ограничения на эффективность преобразования энергии в работу и, кроме того, позволяет ввести критерии возможности самопроизвольного протекания того или иного процесса. Процесс называется самопроизвольным, если он осуществляется без каких-либо воздействий, когда система предоставлена самой себе. Существуют процессы, при которых внутренняя энергия системы не меняется (ДЕ = 0). К таким процессам относится, например, ионизация уксусной кислоты в воде. Целый ряд самопроизвольных процессов протекает с увеличением внутренней энергии (ДЕ > 0). Сюда относятся, в частности, типичные реакции образования бионеорганических соединений альбумина (белок плазмы крови) с ионами металлов, например Сu2+. Изменение внутренней энергии АЕ для закрытых систем не может служить критерием самопроизвольного протекания процессов. Следовательно, первого начала термодинамики, из которого получен этот критерий, недостаточно для решения вопроса о самопроизвольности, равно как и об эффективности процессов. Решение этих вопросов достигается с помощью второго начала термодинамики. Для формулировки второго начала термодинамики необходимо ввести понятия обратимого и необратимого в термодинамическом смысле процессов.Если система находится в равновесии, это состояние поддерживается как угодно долго при неизменности внешних условий. При изменении внешних условий состояние системы может меняться, т. е. в системе может протекать процесс. Процесс называется термодинамически обратимым, если при переходе из начального состояния 1 в конечное состояние 2 все промежуточные состояния оказываются равновесными. Процесс называется термодинамически необратимым, если хоть одно из промежуточных состояний не–равновесно. Обратимый процесс можно осуществить лишь при достаточно медленном изменении параметров системы – температуры, давления, концентрации веществ и др. Скорость изменения параметров должна быть такой, чтобы возникающие в ходе процесса отклонения от равновесия были пренебрежимо малы. Следует отметить, что с обратимостью связана важная проблема медицины – консервация тканей при низких температурах. Обратимые процессы являются предельным случаем реальных процессов, происходящих в природе и осуществляемых в промышленности или в лабораториях.
В качестве критерия самопроизвольности процессов в открытых и закрытых системах вводится новая функция состояния – энергия Гиббса. Эта функция получила название в честь великого американского физика Д. У. Гиббса (1839—1903), который вывел эту функцию, а затем использовал в термодинамических работах.Энергия Гиббса определяется через энтальпию Н и энтропию S с помощью соотношений:
G = H – S, ДG = ДH – ДS.
На основе энергии Гиббса второе начало термодинамики можно сформулировать следующим образом: в изобарно-изотермических условиях (р, Т = const) в системе самопроизвольно могут осуществляться только такие процессы, в результате которых энергия Гиббса системы уменьшается (ДG <0).В состоянии равновесия энергия Гиббса системы не ме–няется (G = const, AG = 0).
ДG < 0, р, Т = const. Из изложенного вытекает, что энергия Гиббса играет большую роль в изучении биоэнергетических процессов. С помощью этой функции состояния можно прогнозировать направление самопроизвольных процессов в биологических системах и рассчитывать мак-симально достижимый КПД.
Энергия Гиббса G так же, как и энтальпия Н, является функцией состояния системы. Поэтому изменение энергии Гиббса ДG может использоваться для характеристики химических превращений аналогично изменению энтальпии ДН. Уравнения реакции, для которых указывается соответствующее этим реакциям изменение энергии Гиббса, также называются термохимическими. Химические реакции, при протекании которых происходит уменьшение энергии Гиббса системы (ДG < 0) и совершается работа, называются экзергоническими. Реакции, в результате которых энергия Гиббса возрастает (ДG > 0) и над системой совершается работа, называются эндергоническими. Выведенная на основе второго начала термодинамики энергия Гиббса является функцией состояния. Следовательно, так же, как и для энтальпии, может быть сформулирован закон Гесса для энергии Гиббса в следующей форме: изменение энергии Гиббса при образовании заданных продуктов из данных реагентов при постоянных давлении и температуре не зависит от числа и вида реакций, в результате которых образуются эти продукты.Важный пример применения закона Гесса – расчет энергии Гиббса реакции окисления глюкозы дикислородом. Изменение энергии Гиббса в этой реакции при р = 101 кПа и Т = 298°К, определенное вне организма, равно ДG° = –2880 кДж/моль. Соответствующее термохимическое уравнение записывается в виде:
С6Н12О6 + 6О2 = 6СО2 + 6Н2О, ДGp-я° = –2880 кДж/моль.
Скорость химических реакций. Зависимость скорости реакции от концентрации. Закон действующих масс. Константа скорости реакции
Под скоростью химической реакции понимается изменение концентрации одного из реагирующих веществ в единицу времени. Концентрация вещества определяется числом молей в литре. При этом безразлично, о каком из участвующих в реакции веществ идет речь: псе они связаны между собой уравнениями реакции. По изменению концентрации одного из веществ можно судить о соответствующих изменениях концентраций всех остальных. При рассмотрении вопроса о скорости реакции необходимо различать гомогенные и гетерогенные реакции.Гомогенные реакции протекают в однородной среде, например в смеси газов или в водном растворе. Например:
2S02 + 02 -> 2SOsгаз газ газ
Гетерогенные реакции идут на поверхности соприкосновения твердого вещества и газа, твердого вещества и жидкости и т. д. Например:
Fe + 2HCl-> FeCl2 + H2t
В связи с этим скорость гомогенной реакции и скорость гетерогенной реакции определяются различно.Скорость гомогенной реакции математически можно отразить так:
V=+,- дельта с/дельта t
v — скорость реакции в гомогенной системе; с,, с2 — начальная и конечная концентрации одного из веществ, измеряемая числом молей в литре (моль/л);г,, t2 — моменты времени (секунды, минуты).Если речь идет о концентрации исходного вещества (концентрация которого с течением времени уменьшается), то берется знак «-». Если скорость оценить увеличением концентрации одного из продуктов реакции, то берется знак «+».Скорость реакции выражается в моль/л-с.Учитывая это, скорость гомогенной реакции можно определить так: Скорость гомогенной реакции определяется количеством вещества, вступающего в реакцию или образующегося при реакции за единицу времени в единице объема. Если реакция протекает между веществами в гетерогенной системе, то реагирующие вещества соприкасаются между собой не по всему объему, а только на поверхности. В связи с этим определение скорости гетерогенной реакции следующее. Скорость гетерогенной реакции определяется числом молей вещества, вступившего в реакцию или образующегося при реакции за единицу времени на единице поверхности. В математической формуле это можно отразить так:
v гетерог= дельта v / S дельта t
v — скорость гетерогенной реакции;
Av — количество молей вещества, вступившего или образующегося в реакции (моль);
S — площадь поверхности соприкосновения (м );
t — промежуток времени (секунда, минута).
Скорость реакции выражается в моль/м –с
Факторы, влияющие на скорость химической реакцииРассмотрим важнейшие факторы, которые влияют на скорость химической реакции. Скорость химической реакции зависит от следующих факторовПрирода реагирующих веществ. Под природой вещества понимают физическую природу, обусловленную строением его молекулы и взаимным влиянием атомов внутри ее, которое зависит от их электронного строения. Например, при равных условиях реакция более активного металла (цинка) с раствором соляной кислоты идет с бурным выделением водорода, менее активного (олово) — довольно медленно:
Zn + 2НС1 4 ZnCL, + Н2Т
Sn + 2HC1 =nCl2 + H2t
Концентрация реагирующих веществ. При увеличении концентрации реагирующих веществ число столкновений между их молекулами увеличивается, поэтому увеличивается и скорость реакции. Например, горение угля в чистом кислороде происходит быстрее, чем в воздухе, где содержание кислорода составляет 20-21%:с + о2 -> со2Площадь поверхности соприкосновения реагирующих веществ. Для твердых веществ скорость прямо пропорциональна поверхности реагирующих веществ, которую можно увеличить при измельчении и перемешивании.
Например, быстрее реагирует с соляной кислотой порошок алюминия, чем кусочек алюминия:
2А1 + 6НС1 =2А1С13 + ЗН2Т
Температура. При повышении температуры скорость большинства реакций увеличивается. Зависимость скорости реакции от температуры определяется правилом Вант-Гоффа: при повышении температуры на 10° скорость реакции увеличивается в 2-4 раза. Например, водород и кислород при комнатной температуре практически не взаимодействуют, а при 600°С реагируют мгновенно (взрыв):2Н2 + 02 Hi 2H20 Увеличение температуры приводит к увеличению числа активных молекул, обладающих достаточной энергией для взаимодействия.Катализатор. Катализаторами называют вещества, которые увеличивают скорости химических реакций, но сами при этом не расходуются. Явление такого ускорения реакций называется катализом. В зависимости от того, в одинаковых или различных агрегатных состояниях находится катализатор и реагирующие вещества, различают гомогенный и гетерогенный катализ. При гетерогенном катализе, когда катализатор представляет собой твердое вещество, а реакция проходит в растворе или газе, реагирующие вещества адсорбируются на поверхности катализатора, где и происходит собственно химическая реакция:
СН2=СН2 + Н2 =СН3-СН3этилен этан
N2 + 3H2=NH3 аммиак
При гомогенном катализе, когда катализатор и реагирующие вещества находятся в одной фазе, катализатор ускоряет реакцию путем образования промежуточных веществ при взаимодействии с одним из исходных компонентов, например:
2SO, + О., =2SO, (NO, - катализатор)
2 2 3 2
Все вещества в газовой фазе.
СН3СООН + С2Н2ОН -> СН3СООС2Н5 + Н20
уксусная этанол 2 4 этиловый эфир. кислота уксусной кислоты
Все реагенты в жидкой фазе.Особую роль играют биологические катализаторы — ферменты. При их участии протекают сложные химические процессы в растительных и животных организмах. Однако, кроме катализаторов, которые ускоряют реакции, есть вещества, которые замедляют химические реакции, то есть снижают скорость реакции. Их называют ингибиторами. Например соединения мышьяка, ртути, свинца, цианистые соединения и др.Влияние концентрации реагирующих веществ может быть объяснено из представлений, согласно которым химическое взаимодействие является результатом столкновения частиц. Увеличение числа частиц в заданном объеме приводит к более частым их столкновениям, т.е. к увеличению скорости реакции. Количественно зависимость между скоростью реакции и молярными концентрациями реагирующих веществ описывается основным законом химической кинетики — законом действующих масс. Скорость химической реакции при постоянной температуре прямо пропорциональна произведению концентраций реагирующих веществ.Для мономолекулярной реакции скорость реакции определяется концентрацией молекул вещества А:v = k*[A]где k — коэффициент пропорциональности, который называется константой скорости реакции; [А] — молярная концентрация вещества А.В случае бимолекулярной реакции, ее скорость определяется концентрацией молекул не только вещества А, но и вещества В:v = k*[A]*[B]В случае тримолекулярной реакции, скорость реакции выражается уравнением: v = k*[A]2*[B]В общем случае, если в реакцию вступают одновременно т молекул вещества А и n молекул вещества В, т. е.тА + пВ = С, уравнение скорости реакции имеет вид:v = k*[A]m*[B]nЭто уравнение есть математическое выражение закона действующих масс в общем виде. Чтобы понять физический смысл константы скорости реакции, надо принять в написанных выше уравнениях, что [А] = 1 моль/л и [В] = 1 моль/л (либо приравнять единице их произведение), и тогда v = k. Отсюда ясно, что константа скорости k численно равна скорости реакции, когда концентрации реагирующих веществ (или их произведение в уравнениях скорости) равны единице. Общее выражение для скорости химической реакции получено для данной, фиксированной температуры. В общем же случае, поскольку скорость реакции зависит от температуры, закон действующих масс записывается как v(T) = k(T) *[A]m*[B]n де v и k являются функциями температуры Константа скорости химической реакции, ее зависимость от температуры Многочисленные опыты показывают, что при повышении температуры скорость большинства химических реакций существенно увеличивается, причем для реакций в гомогенных системах при нагревании на каждые десять градусов скорость реакции возрастает в 2—4 раза (правило Вант-Гоффа).Это правило связано с понятием температурного коэффициента скорости реакции г и определяется соотношением г = kТ+10 / kТ Значение температурного коэффициента г дает возможность рассчитать изменение скорости реакции при увеличении температуры на некоторое число градусов от Т1 до Т2 по формуле v(Т1)/v(Т2) = г(Т2-Т1)/10Очевидно, что при повышении температуры в арифметической прогрессии скорость реакции возрастает в геометрической.
Похожие работы
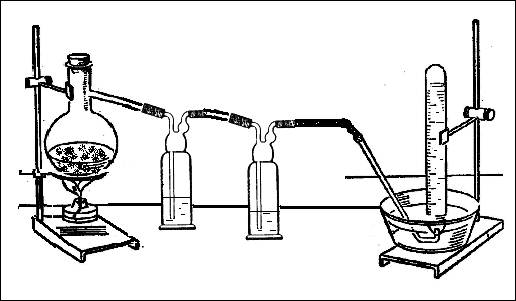
... подкрепляет своим одобрением неправильный или не вполне точный ответ ученика. 1.2 Совершенствование школьного химического эксперимента при проблемном обучении 1.2.1 Принципы разработки методической системы и содержания опытов по химии в системе проблемного обучения Характерной особенностью развивающего обучения является широкое использование проблемного подхода, который включает создание ...
ависимо от способа получения и места нахождения. 2. Строение внешнего электронного уровня атома калия и кальция. 1 правило Клечковского. Строение внешнего электронного уровня атома скандия. 2правило Клечковского У атома аргона остаются незанятыми все орбитали 3d-подуровня. Однако у следующих за аргоном элементов – калия и кальция – заполнение 3-го электронного слоя временно прекращается, и ...
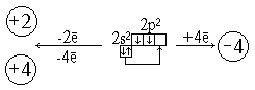
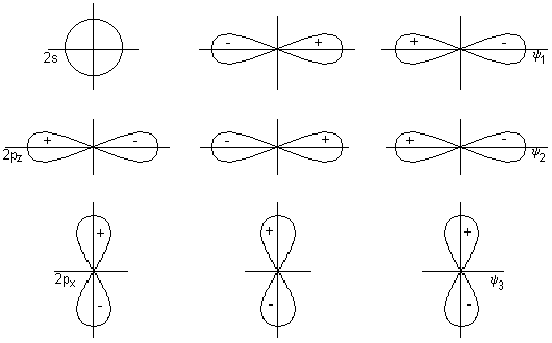
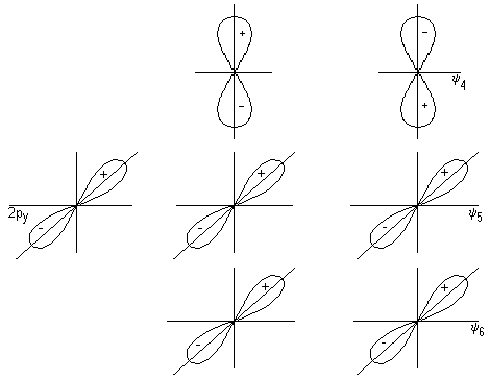
... разовая) – 0,01%. 4 Содержание Введение......................................................................................................................4 Глава 1. Межпредметные связи в курсе школьного предмета химии на примере углерода и его соединений.......................................................................5 1.1 Использование межпредметных связей для формирования у учащихся ...
... учреждение страны, а в ее задачи входило усовершенствование наук, просвещение, а также усовершенствование мануфактур, ремесел и фабрик. В то же время в начале XIX столетия, особенно после Отечественной войны 1812 г., в развитии химии в России появились новые черты. Смена мануфактурного производства фабрично-заводским выдвинула перед учеными множество практических задач, связанных с рациональной ...
0 комментариев