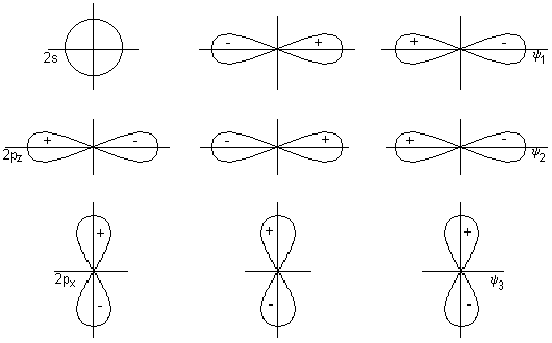
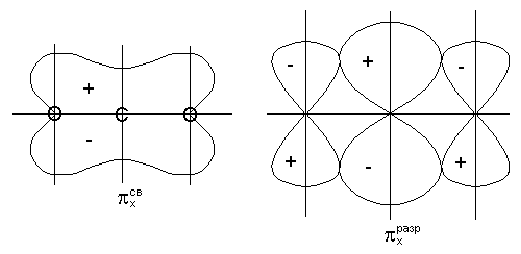
Ковалентная
(неполярная,
полярная) связь.
Механизмы
образования
ковалентной
связи
Году американский
ученый Льюис
высказал
предположение
о том, что химическая
связь образуется
за счет обобществления
двух электронов
Скорость
химических
реакций. Порядок
и молекулярность
реакций
Зависимость
скорости реакции
от температуры.
Правило Вант-Гоффа.
Уравнение
Аррениуса
Энергия
активации.
Активированные
комплексы.
Уравнение
Аррениуса
Растворы.
Физическая
и химическая
теории растворов
Фазовые
равновесия
в гетерогенных
системах, фазовые
превращения
и правило фаз.
Диаграммы
состояния
Сильные
и слабые электролиты.
Константа
диссоциации.
Закон разбавления
Оствальда.
Слабые электролиты.
Константа
диссоциации
Электролитическая
диссоциация
воды. Ионное
произведение
воды. Водородный
показатель
среды. Понятие
об индикаторах
Гидролиз
солей. Обратимый
и необратимый
(полный) гидролиз.
Роль процессов
гидролиза при
эксплуатации
котельных
установок.ъ
Растворимость
веществ. Произведение
растворимости.
Механизм
накипеообразования
Концентрации
или парциальные
давления окисленной
и восстановленной
форм
Окислитель
или восстановитель
иногда дополнительно
расходуется
на связывание
получающихся
продуктов
(солеобразование).Например,
в реакции
Электродный
потенциал.
Влияние температуры
и концентрации
на величину
электродного
потенциала.
Уравнение
Нернста
Практическое
использование
электрохимических
процессов.
Химические
источники тока
Коррозия
металлов. Основные
виды коррозии.
Химическая
коррозия
Коррозия
металлов.
Электрохимическая
коррозия
Методы защиты
металлов от
коррозии: изменение
свойств коррозионной
среды, защитные
покрытия,
электрохимическая
защита
Распространенность
химических
элементов.
Основные классы
неорганических
соединений
Навигация
Фазовые равновесия в гетерогенных системах, фазовые превращения и правило фаз. Диаграммы состояния
Общая и неорганическая химия
442397
знаков
6
таблиц
13
изображений
21. Фазовые равновесия в гетерогенных системах, фазовые превращения и правило фаз. Диаграммы состояния
Фазовое равновесие, сосуществование термодинамически равновесных фаз гетерогенной системы. Является одним из основных случаев термодинамического равновесия и включает в себя условия равенства температуры всех частей системы (термическое равновесие), равенства давления во всем объеме системы (механическое равновесие) и равенство химических потенциалов каждого компонента во всех фазах системы, что обеспечивает равновесное распределение компонентов между фазами. Число фаз f, находящихся одновременно в равновесии, связано с числом компонентов k, числом n независимых параметров, определяющих состояние системы (обычно, когда учитывается только влияние температуры и давления, n = 2), и числом термодинамических степеней свободы v уравнением: v = k + 2 - f (см. Фаз правило).В общем виде условие фазовое равновесие, согласно принципу равновесия Гиббса, сводится к максимуму энтропии S системы при постоянстве внутренней энергии U, общего объема V и числа молей каждого компонента ni-. Этот принцип можно выразить также как условие минимума любого из термодинамических потенциалов: внутренней энергии U, энтальпии H, энергии Гиббса G, энергии Гельмгольца А при условии постоянства соответствующих параметров состояния, включая число молей каждого компонента.Фазовые равновесия могут быть стабильными и метастабильными. Те и другие являются локально устойчивыми, то есть устойчивыми по отношению к малым возмущениям параметров состояния - температуры, давления, состава (концентраций компонентов). Метастабильные фазовые равновесия отличаются тем, что они неустойчивы к некоторым конечным изменениям этих параметров, ведущим, в частности, к переходу к другим фазам. Например, пересыщенный раствор или переохлажденный расплав неустойчивы по отношению к кристаллической фазе. Поскольку метастабильное состояние системы локально устойчиво, переход к стабильному состоянию требует преодоления некоторого активационного барьера и протекания процесса зародышеобразования (см. Зарождение новой фазы).
Следует отметить некоторые особенности метастабильных фаз: при одной и той же температуре давление пара выше над метастабильной фазой, чем над стабильной; при одном и том же давлении температура плавления метастабильной фазы ниже, чем стабильной; растворимость метастабильной фазы при постоянных давлении и температуре выше, чем стабильной. Последнее справедливо как для жидких, так и для твердых растворов.Критерий достижения фазового равновесия. Наиболее общий критерий достижения фазового равновесия - сходимость значений CB-B системы при их измерении, если подходить к состоянию фазового равновесия сверху (со стороны более высоких температур) и снизу (со стороны низких температур). Достижение фазового равновесия или хотя бы приближение к нему - важнейший вопрос при изучении диаграмм состояния, в том числе диаграмм растворимости, диаграмм плавкости, диаграмм давления пара, а также в физико-химическом анализе. При исследовании растворимости для достижения фазового равновесия применяют длительную (от нескольких часов до нескольких месяцев) выдержку образца с перемешиванием в термостате. В случае образования в системе твердых растворов рекомендуется подход к равновесию сверху, от более высоких температур, сочетающий быстрое охлаждение с целью получения мелких кристаллов и интенсивное перемешивание. При исследовании систем методом термического анализа обычно используют образцы, полученные сплавлением компонентов с последующим медленным охлаждением. В случае образования в системе твердых растворов и инконгруэнтно плавящихся фаз, а также фаз, разлагающихся в твердом состоянии, требуется проведение предварительного отжига образца при фиксированной температуре - от нескольких часов до нескольких месяцев. Для ускорения отжига сплавленных образцов рекомендуется предварительное быстрое охлаждение расплава.
При изучении твердых тел. состоящих из тугоплавких или разлагающихся при высоких температурах компонентов, применяют такие методы подготовки образцов, как прессование таблеток смесей перед отжигом и промежуточное перетирание смесей при отжиге, отжиг смесей солей или гелей, осажденных из водных или других растворов и т. п.Типы фазовых равновесий. В однокомпонентной системе (при наличии полиморфных превращений) возможны 4 вида двухфазных равновесий: жидкость - пар, кристалл - пар, кристалл - жидкость и кристалл - кристалл; 4 вида трехфазных равновесий: кристалл - жидкость - пар, кристалл - кристалл - жидкость, кристалл - кристалл - пар и кристалл - кристалл - кристалл; при этом не учитывается возможность образования жидких кристаллов. В двойных системах (компоненты А и В) возможны те же виды двухфазных равновесий, но число возможных видов трехфазных равновесий достигает 26 вследствие того, что играет роль не только природа сосуществующих фаз (их агрегатное состояние), но и взаимное расположение фазовых полей на диаграмме состояния в координатах температура - состав (давление предполагается постоянным). Все эти фазовые равновесия делятся на два типа: эвтектическое фазовое равновесие, при которых из трех одновременно участвующих в равновесии фаз при понижении температуры одна испытывает превращение, а две другие при этом образуются, и перитектическое фазовое равновесие, когда две фазы взаимодействуют (превращаются), при этом образуется третья фаза.
В простейшем случае, если на основе компонентов А и В возможно образование жидкого раствора L и двух твердых растворов a и b, эвтектического и перитектического фазовых равновесий можно записать соответственно в виде реакций:
![]()
Поскольку в двойной системе состояние трехфазного равновесия является нонвариантным, эвтектические и перитектические реакции происходят при постоянной температуре, называемой соотв. эвтектической или перитектической, то есть на диаграмме состояния этим равновесиям отвечают горизонтали. В случае, если в определенной области температур и составов все три равновесно сосуществующие фазы являются твердыми (у одного из компонентов существуют полиморфные модификации с образованием твердого раствора g), возможны трехфазные равновесия, называют эвтектоидными и перитектоидными. Их можно представить соответствующими реакциями, аналогично эвтектическим и перитектическим фазовым равновесиям:
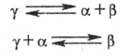
При наличии в некотором температурно-концентрационном интервале двух жидких фаз L1 и L2 и одной твердой (напр., а) возможны трехфазные равновесия, называют монотектическое и синтектическое:
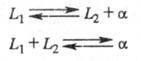
Некоторые виды трехфазных равновесий, например те, при которых образуется жидкость в результате взаимодействием двух кристаллических фаз при понижении температуры, теоретически возможны, но реально, по-видимому, не наблюдаются. При переходе к тройным и более сложным системам число видов многофазных фазовых равновесий возрастает еще больше (см. Тройная точка).Распределение компонентов между фазами системы при фазовом равновесии описывается законом распределения, устанавливающим, что отношение термодинамических активностей примеси в двух фазах при фазовое равновесие является постоянной величиной. В первом приближении активности компонентов можно заменить их концентрациями. Одним из условий выполнимости закона распределения вещества между фазами является одинаковость молекулярного состояния растворенного вещества в обеих фазах, то есть отсутствие ассоциации молекул. Замена активностей на концентрации допустима, если коэффициент активности компонента в обеих фазах не зависят от концентрации, то есть для идеальных растворов (это условие обычно выполняется для очень разбавленных растворов, в случае микроконцентраций). Отношение активностей компонентов называют коэффициентом распределения или коэффициентом относительной летучести и т. п. Частные случаи закона распределения - правила и законы, выражающие равновесное распределение вещества в двухфазных системах. Например, для расчета равновесия жидкости и пара пользуются законами Рауля и Генри, первым - для вещества, находящегося в избытке, вторым - для вещества, являющегося примесью. Распределение растворенного вещества между двумя несмешивающимися жидкостями при постоянной температуре характеризуется тем, что отношение его концентраций в этих двух фазах сохраняется постоянным (закон Бертло - Нернста). Распределение примеси между жидкой и твердой кристаллической фазой описывается распределениями Хлопина (равновесия) и Дёрнера-Хоскинса.Законы распределения являются основой разнообразных гетерогенных методов очистки (разделения), хотя само фазовое равновесие в процессе проведения этих методов очистки достигается далеко не всегда, а иногда сама возможность очистки обусловлена отсутствием фазового равновесия (см. Кристаллизационные методы разделения смесей, Ректификация, Экстракция жидкостная).
Диаграммы состояния
Диаграммы состояния, или диаграммы фазового равновесия в удобной графической форме показывают фазовый состав сплава в зависимости от температуры и концентрации. Диаграммы состояния строят для условий равновесия (окончательное состояние). Равновесное состояние соответствует минимальному значению свободной энергии. Это состояние может быть достигнуто только при очень малых скоростях охлаждения или длительном нагреве. Однако истинное равновесие достигается редко, наиболее часто системы находятся в метастабильном состоянии (неустойчивом), и под воздействием внешних факторов могут переходить в другие более устойчивые состояния. Метастабильные состояния нередко сообщают сплавам высокие механические и другие свойства.
Правило фаз
Диаграммы фазового равновесия характеризуют окончательное состояние сплавов, то есть после того как все превращения в них произошли и полностью закончились. Это состояние зависит от внешних условий (Т0 С; Р, МПа) и характеризуется числом и концентрацией образовавшихся фаз. Закономерность изменения числа фаз в гетерогенной системе определяется правилом фаз.
Правило фаз устанавливает зависимость между числом степеней свободы, числом компонентов и числом фаз и выражается уравнением
С = К - Ф + 2,
где С - число степеней свободы системы (или вариантность);
К - число компонентов, образующих систему;
2 - число внешних факторов (Т и Р);
Ф - число фаз, находящихся в равновесии.
Под числом степеней свободы (вариантностью системы) понимают возможность изменения температуры, давления и концентрации без изменения числа фаз, находящихся в равновесии.
При нормальных условиях изменяется только один фактор -Т0С, Р =const, тогда:
С=К - Ф + 1.
Число степеней свободы не может быть меньше нуля, тогда К-Ф+1>0, а Ф<К+1, то есть число фаз в сплаве, не может быть больше чем число компонентов плюс единица. Таким образом, в двойной системе может быть не более трёх фаз.При С = 0 - существует в равновесии сразу три фазы - имеется нонвариантное равновесие (безвариантное). При таком равновесии сплав может существовать только при условии - постоянная температура и определённый состав всех фаз, находящихся в равновесии. То есть кристаллизация (или превращение) начинается и заканчивается при постоянной температуре. Если С =1 или 2, то кристаллизация или превращение протекает с течением времени в интервале температур.
Построение диаграмм состояния
Диаграмма состояния показывает изменение состояния сплавов в зависимости от температуры (P = const) и концентрации.
Если в системе имеется два компонента, то диаграмма будет иметь два измерения: первое - температурная шкала, второе - концентрация сплава (рисунок 2).
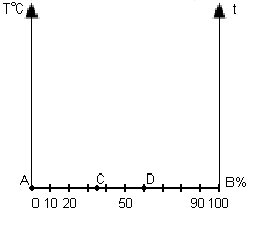
Рисунок 2.
Каждая точка на оси абсцисс соответствует определённому содержанию каждого компонента. Общее содержание компонентов в сплаве - 100%.
Крайние ординаты на диаграмме соответствуют чистым компонентам, а ординаты между ними - двойным сплавам.
Через точку С проходит сплав содержащий 35% компонента В и, соответственно, 65% компонента А.
Каждая точка на диаграмме состояния показывает состояние сплава данной концентрации при данной температуре. Каждая вертикаль соответствует изменению температуры определенного сплава. Изменение фазового состояния сплава отмечается на диаграмме точкой.
Линии, соединяющие точки аналогичных превращений, разграничивают на диаграмме области аналогичных фазовых состояний.
Вид диаграммы состояния зависит от того, как реагируют оба компонента друг с другом в твердом и жидком состоянии, то есть, растворимы ли они в жидком и твердом состоянии, образуют ли химические соединения и так далее.
Обычно диаграммы состояния строят, экспериментально используя термический анализ, то есть строят кривые охлаждения и по остановкам и перегибам на этих кривых, вызванным тепловым эффектом превращений, определяют температуры превращений. Эти температуры называют критическими точками.
Температуру металлов измеряют обычно при помощи термопары.
Имея достаточное количество сплавов, и определив в каждом сплаве температуры превращений, можно построить диаграмму состояния.
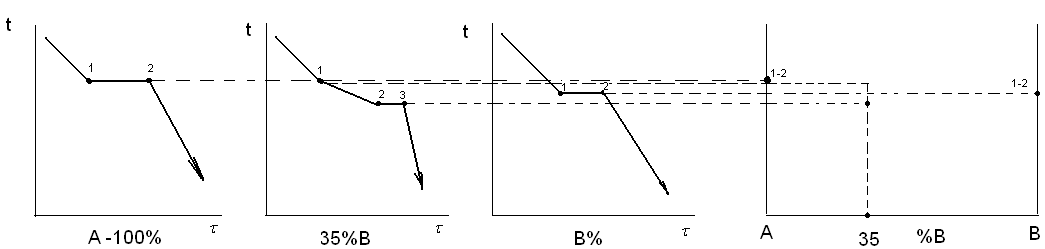
Рисунок 3. Построение кривых охлаждения
Диаграмма состояния показывает, какую структуру будет иметь медленно охлажденный сплав при любой температуре.
Растворы как многокомпонентные системы. Способы выражения состава растворов. Молярная доля, массовая доля. Молярная концентрация, молярная концентрация эквивалентов, моляльная концентрация
Растворы – это однородные (гомогенные) дисперсные системы, состоящие из двух или большего числа компонентов (относительные количества которых могут меняться в широких пределах) и продуктов их взаимодействия.
Растворы занимают промежуточное положение между механическими смесями (растворы характеризуются непостоянством своего состава и могут быть разделены на составные части) и химическими соединениями (растворы однородны, устойчивы, образование растворов сопровождается энергетическим эффектом).
В настоящее время установлено, что при растворении молекулы растворяемого вещества связываются с молекулами растворителя, при этом образуются сольваты (если растворитель вода, то образуются гидраты). На разрушение связей между молекулами энергия затрачивается, а при образовании гидрата (сольвата) энергия выделяется; разница между этими энергиями будет наблюдаться в виде теплового эффекта растворения, которая может быть как положительной, так и отрицательной.
Способы выражения концентрации растворов.
1) Массовая доля раствора щ (х). Выражается отношением массы растворенного вещества m(х) к массе раствора.
![]()
Является величиной безразмерной или выражается в процентах:
![]()
Например, 15%-ный раствор: массовая доля щ (х) = 0,15
2) Молярная концентрация раствора С(х). Выражается отношением количества растворенного вещества n(x) к объему раствора, выраженному в литрах.
![]()
Т.к. количество вещества n(x) выражается отношением массы вещества m(x) к его молярной массе M(x), то молярную концентрацию раствора удобно выразить как
![]()
Концентрация — величина, характеризующая количественный состав раствора.
Согласно правилам ИЮПАК, концентрацией растворённого вещества (не раствора) называют отношение количества растворённого вещества или его массы к объёму раствора (моль/л, г/л), то есть это соотношение неоднородных величин.
Те величины, которые являются отношением однотипных величин (отношение массы растворённого вещества к массе раствора, отношение объёма растворённого вещества к объёму раствора) правильно называть долями. Однако на практике для обоих видов выражения состава применяют термин концентрация и говорят о концентрации растворов.
Существует много способов выражения концентрации растворов. Массовая доля (также называют процентной концентрацией)
Массовая доля — отношение массы растворённого вещества к массе раствора. Массовая доля измеряется в долях единицы.
![]()
где:
m1 — масса растворённого вещества, г ;
m — общая масса раствора, г.
Массовое процентное содержание компонента, m%
m%=(mi/Уmi)*100
Молярность (молярная объёмная концентрация)
Молярная концентрация — количество растворённого вещества (число молей) в единице объёма раствора. Молярная концентрация в системе СИ измеряется в моль/мі, однако на практике её гораздо чаще выражают в моль/л или ммоль/л. Также распространено выражение в «молярности». Возможно другое обозначение молярной концентрации CM, которое принято обозначать М. Так, раствор с концентрацией 0,5 моль/л называют 0,5-молярным. Примечание: термин «моль» не склоняется по падежам. После цифры пишут «моль», подобно тому, как после цифры пишут «см», «кг» и т. д.
![]() где:
где:
н — количество растворённого вещества, моль;
V — общий объём раствора, л.
Эквивалентная концентрация сeqB растворенного вещества В – это отношение эквивалентного количества вещества neqB к объему раствора:
сeqB = neqB /V(p).
Единица эквивалентной концентрации – моль/л.
Раствор, в котором эквивалентная концентрация растворенного вещества равна сeqB (моль/л), характеризуется нормальноcтью, численно равной значению сeqB; обозначение нормальности раствора – н. (поcле числа). Например, раствор с эквивалентной концентрацией серной кислоты сeq(H2SO4) = 1 моль/л. может быть обозначен как 1 н. H2SO4 (однонормальный раствор серной кислоты в воде).
Эквивалентная концентрация растворенного вещества и нормальность раствора определяются эквивалентным количеством растворенного вещества и, следовательно, как и последнее, зависят от эквивалентного числа, постоянного только для конкретной реакции, причем сeqB = zBсB
Моляльная концентрация Cml (моль/кг) - это отношение количества растворенного вещества n к массе растворителя msol:
Cml = n
-----
msol
Моляльная концентрация не зависит от температуры раствора, т.к. масса раствора при различных температурах остается постоянной; а объем раствора, изменяется с изменением его температуры.
Обратите внимание, на то, что несмотря на созвучность в названиях: молярная и моляльная - это различные способы выражения концентрации растворенного вещества в растворе
Способы выражения состава раствора Важнейшей характеристикой раствора является его состав. Состав определяет раствор как в качественном отношении (из каких компонентов состоит раствор), так и в количе-ственном (в каких относительных количествах тот или иной компонент содержится в растворе).Существует много способов выражения количественного состава. Рассмотрим основные из них.
1. Молярная доля(хi) - отношение количества вещества (моль) компонента ni, содержащегося в данной системе, к общему числу молей в системе Eni.
Xi =ni/Еni. (11.1)
2. Объемная доля (фi) - отношение объема компонента (Vi), содержащегося в систе-ме, к общему объему V:
(Pi = Vi,/ V (11.2)
3. Массовая доля (a>i) - отношение массы компонента mi к массе смеси Emi.
coi = mi /Emi. (11.3)
4.Молярная концентрация (ci) - отношение количества вещества ni (моль), содержащегося в системе, к объему этой системы V:
Ci = ni/ V (11.4)
5.Молярностъ растворенного вещества (bi) - отношение количества растворенно-го компонента ni к массе вещества растворителя mp. Обычно рассматривают содержание в 1000 г растворителя.(bi) = ni /mp [моль/кг] (11.5)
6.Молярная концентрация эквивалента (cif) - отношение количества эквивалентоврастворенного вещества nif к объему системы V
ci = nif / V (11.6)
Свойства растворов неэлектролитов. Явление осмоса. Закон Вант-Гоффа. Давление насыщенного пара растворителя над раствором. Первый и второй законы Рауля
Первый закон Рауля связывает давление насыщенного пара над раствором с его составом; он формулируется следующим образом:
Парциальное
давление насыщенного
пара компонента
раствора прямо
пропорционально
его мольной
доле в растворе,
причём коэффициент
пропорциональности
равен давлению
насыщенного
пара над чистым
компонентом.
![]() Для бинарного
раствора, состоящего
из компонентов
А и В (компонент
А считаем
растворителем)
удобнее использовать
другую формулировку:
Для бинарного
раствора, состоящего
из компонентов
А и В (компонент
А считаем
растворителем)
удобнее использовать
другую формулировку:
Относительное понижение парциального давления пара растворителя над раствором не зависит от природы растворённого вещества и равно его мольной доле в растворе.
![]()
Растворы, для которых выполняется закон Рауля, называются идеальными. Идеальными при любых концентрациях являются растворы, компоненты которых очень близки по физическим и химическим свойствам (оптические изомеры, гомологи и т. п.), и образование которых не сопровождается изменением объёма и выделением либо поглощением теплоты. В этом случае силы межмолекулярного взаимодействия между однородными и разнородными частицами примерно одинаковы, и образование раствора обусловлено лишь энтропийным фактором. Отклонения от закона РауляРастворы, компоненты которых существенно различаются по физическим и химическим свойствам, подчиняются закону Рауля лишь в области очень малых концентраций; при больших концентрациях наблюдаются отклонения от закона Рауля. Случай, когда истинные парциальные давления паров над смесью больше, чем вычисленные по закону Рауля, называют положительными отклонениями. Противоположный случай, когда парциальные давления паров компонентов оказываются меньше вычисленных — отрицательные отклоненияПричиной отклонений от закона Рауля является то обстоятельство, что однородные частицы взаимодействуют друг с другом иначе, чем разнородные (сильнее в случае положительных и слабее в случае отрицательных отклонений).Реальные растворы с положительными отклонениями от закона Рауля образуются из чистых компонентов с поглощением теплоты (ДНраств > 0); объём раствора оказывается больше, чем сумма исходных объёмов компонентов (ДV > 0). Растворы с отрицательными отклонениями от закона Рауля образуются с выделением теплоты (ДНраств < 0); объём раствора в этом случае будет меньше, чем сумма исходных объёмов компонентов (ДV < 0).
Второй закон Рауля
Тот факт, что давление паров над раствором отличается от давления паров над чистым растворителем, существенно влияет на процессы кристаллизации и кипения. Из первого закона Рауля выводятся два следствия, касающиеся понижения температуры замерзания и повышения температуры кипения растворов, которые в объединённом виде известны как второй закон Рауля. Понижение температуры кристаллизации растворов
Условием кристаллизации является равенство давления насыщенного пара растворителя над раствором давлению пара над твёрдым растворителем. Поскольку давление пара растворителя над раствором всегда ниже, чем над чистым растворителем, это равенство всегда будет достигаться при температуре более низкой, чем температура замерзания растворителя. Так, океанская вода начинает замерзать при температуре около минус 2 °C.
Разность между температурой кристаллизации растворителя T°fr и температурой начала кристаллизации раствора Tfr есть понижение температуры кристаллизации.
Понижение температуры кристаллизации бесконечно разбавленных растворов не зависит от природы растворённого вещества и прямо пропорционально моляльной концентрации раствора.
![]()
Поскольку по мере кристаллизации растворителя из раствора концентрация последнего возрастает, растворы не имеют определённой температуры замерзания и кристаллизуются в некотором интервале температур. Повышение температуры кипения растворов
Жидкость кипит при той температуре, при которой общее давление насыщенного пара становится равным внешнему давлению. Если растворённое вещество нелетуче (то есть давлением его насыщенных паров над раствором можно пренебречь), то общее давление насыщенного пара над раствором равно парциальному давлению паров растворителя. В этом случае давление насыщенных паров над раствором при любой температуре будет меньше, чем над чистым растворителем, и равенство его внешнему давлению будет достигаться при более высокой температуре. Таким образом, температура кипения раствора нелетучего вещества Tb всегда выше, чем температура кипения чистого растворителя при том же давлении T°b.
Повышение температуры кипения бесконечно разбавленных растворов нелетучих веществ не зависит от природы растворённого вещества и прямо пропорционально моляльной концентрации раствора
![]() .
.
РАСТВОРЫ НЕЭЛЕКТРОЛИТОВ, бинарные или многокомпонентные мол. системы, состав к-рых может изменяться непрерывным образом (по крайней мере, в нек-рых пределах). В отличие от растворов электролитов, в растворах неэлектролитов (мол. р-рах) заряженные частицы в сколько-нибудь заметных концентрациях отсутствуют. Растворы неэлектролитов могут быть твердыми, жидкими и газообразными. В данной статье рассматриваются жидкие р-ры; см. также Твердые растворы.Взаимная р-римость двух жидкостей при заданных т-ре Т и давлении р м. б. полной (неограниченной) или ограниченной. В последнем случае р-ры в нек-рой области составов расслаиваются, т. е. разделяются на две жидкие фазы, отличающиеся по концентрации. В многокомпонентных расслаивающихся р-рах число сосуществующих жидких фаз м. б. более двух. Если один (или более) из компонентов раствора неэлектролитов в чистом состоянии при заданных Т и р является газом или твердым телом, область существования раствора неэлектролитов простирается от чистой жидкости (смеси жидкостей), выступающей в роли р-рителя, до состава, отвечающего насыщ. р-ру.Растворы неэлектролитов служат средой, в к-рой протекают многие прир. и пром. процессы. Изучение и прогнозирование св-в этих систем тесно связаны с такими практич. проблемами, как подбор р-рителей для реализации технол. процессов, получение систем с заданными св-вами, разделение прир. и пром. смесей (включая газы и нефти), глубокая очистка в-в.
Физ. химия изучает широкий диапазон св-в р-ров. Наиб. разработана и имеет практически важные применения равновесная термодинамика р-ров; дальнейший материал посвящен в осн. этому разделу физ. химии р-ров. Кроме того, изучаются транспортные св-ва р-ров-диффузия, теплопроводность, вязкость (см. Физико-химическая гидродинамика), а также спектроскопич., электрич., акустич. и др. физ. св-ва. Методы исследования макроскопич. св-в растворов неэлектролитов и их структурных характеристик во многом аналогичны методам исследования индивидуальных жидкостей, но осн. внимание уделяется рассмотрению концентрац. зависимостей св-в. Важнейшая задача физ.-хим. исследований - установление связи между наблюдаемыми на опыте св-вами, структурой р-ров и характеристиками межмолекулярных взаимодействий. Эксперим. информацию о структуре р-ров и межмолекулярных взаимод. в них дают методы оптической и радиоспектроскопии, дифракционные, электрич. и др. Важную роль в изучении растворов неэлектролитов играет физико-химический анализ, основанный на построении и исследовании фазовых диаграмм, концентрац. зависимостей термодинамич. и др. физ. св-в (показателя преломления, вязкости, теплопроводности, акустич. характеристик и др.). При этом одна из главных задач состоит в том, чтобы на основании анализа диаграмм состав - свойство устанавливать факт образования хим. соединений между компонентами растворов неэлектролитов и находить их характеристики.Значит. влияние на физ. св-ва р-ров (в частности, на рассеяние света) оказывают флуктуации плотности, концентрации, ориентации молекул. Роль флуктуации концентрации особенно велика вблизи критич. точки р-римости (см. Критические явления).Концентрационные зависимости термодинамических функций. Особенностью термодинамич. описания растворов неэлектролитов по сравнению с чистыми компонентами является наличие дополнит. термодинамич. степеней свободы системы, связанных с возможностью изменения состава системы (см. Фаз правило). Число степеней свободы гомогенного n-компонентного р-ра равно n+1. В качестве переменных, определяющих его состояние, наиб. удобно выбрать давление р, т-ру Т и концентрации п — 1 компонентов. Состав растворов неэлектролитов чаще всего выражают через молярные доли компонентов xi, считая независимыми переменными молярные доли всех компонентов, кроме n-го x1,..., xn-1. Для задания концентрации используют и др. шкалы (молярности с, моляльности т).
При описании концентрац. зависимостей термодинамич. ф-ций важную роль играют парциальные молярные величины Mi для i-го компонента, определяемые соотношением:
![]()
где М-любая экстенсивная термодинамич. ф-ция (объем V, внутр. энергия U, энтальпия H, энтропия S, энергии Гельм-гольца и Гиббса F и G, теплоемкость Ср и т.д.), mi-число молей. Важнейшая парциальная молярная величина -химический потенциал mi (парциальная молярная энергия Гиббса); именно через хим. потенциалы формулируются условия хим. и фазового равновесий в системе.
Концентрац.
зависимость
термодинамич.
св-в растворов
неэлектролитов
нередко характеризуют
функциями
смешения Мт
- изменением
термодинамич.
ф-ции М при
образовании
р-ра из чистых
жидкостей.
Рассматривают
смешение при
изотермо-изобар-ных
(Т, р = const) или изотермо-изохорных
(Т, V = const) условиях,
причем наиб.
практич. интерес
представляет
случай Т, р —
const. Молярная ф-ция
смешения при
этих условиях
(![]() )
определена
соотношением:
)
определена
соотношением:
![]()
где
![]()
молярное значение ф-ции M для чистой жидкости i при заданных Т и р. В частности, молярная энергия Гиббса смешения
![]()
где
![]() хим. потенциал
чистой жидкости
i при заданных
Т и р. Для чистых
жидкостей
хим. потенциал
чистой жидкости
i при заданных
Т и р. Для чистых
жидкостей ![]()
О́смос (от греч. ὄумпт «толчок, давление») — процесс односторонней диффузии через полупроницаемую мембрану молекул растворителя в сторону большей концентрации
Явление осмоса наблюдается в тех средах, где подвижность растворителя больше подвижности растворённых веществ. Важным частным случаем осмоса является осмос через полупроницаемую мембрану. Полупроницаемыми называют мембраны, которые имеют достаточно высокую проницаемость не для всех, а лишь для некоторых веществ, в частности, для растворителя. (Подвижность растворённых веществ в мембране стремится к нулю). Если такая мембрана разделяет раствор и чистый растворитель, то концентрация растворителя в растворе оказывается менее высокой, поскольку там часть его молекул замещена на молекулы растворенного вещества (см. Рис. 1). Вследствие этого, переходы частиц растворителя из отдела, содержащего чистый растворитель, в раствор будут происходить чаще, чем в противоположном направлении. Соответственно, объём раствора будет увеличиваться (а концентрация уменьшаться), тогда как объём растворителя будет соответственно уменьшаться.
Например, к яичной скорлупе с внутренней стороны прилегает полупроницаемая мембрана: она пропускает молекулы воды и задерживает молекулы сахара. Если такой мембраной разделить растворы сахара с концентрацией 5 и 10 % соответственно, то через нее в обоих направлениях будут проходить только молекулы воды. В результате в более разбавленном растворе концентрация сахара повысится, а в более концентрированном, наоборот, понизится. Когда концентрация сахара в обоих растворах станет одинаковой, наступит равновесие. Растворы, достигшие равновесия, называются изотоническими.
Осмос, направленный внутрь ограниченного объёма жидкости, называется эндосмосом, наружу — экзосмосом. Перенос растворителя через мембрану обусловлен осмотическим давлением. Оно равно избыточному внешнему давлению, которое следует приложить со стороны раствора, чтобы прекратить процесс, то есть создать условия осмотического равновесия. Превышение избыточного давления над осмотическим может привести к обращению осмоса — обратной диффузии растворителя.В случаях, когда мембрана проницаема не только для растворителя, но и для некоторых растворённых веществ, перенос последних из раствора в растворитель позволяет осуществить диализ, применяемый как способ очистки полимеров и коллоидных систем от низкомолекулярных примесей, например электролитов.
Значение осмосаОсмос играет важную роль во многих биологических процессах. Мембрана, окружающая нормальную клетку крови, проницаема лишь для молекул воды, кислорода, некоторых из растворенных в крови питательных веществ и продуктов клеточной жизнедеятельности; для больших белковых молекул, находящихся в растворенном состоянии внутри клетки, она непроницаема. Поэтому белки, столь важные для биологических процессов, остаются внутри клетки.
Осмос участвует в переносе питательных веществ в стволах высоких деревьев, где капиллярный перенос не способен выполнить эту функцию.Осмос широко используют в лабораторной технике: при определении молярных характеристик полимеров, концентрировании растворов, исследовании разнообразных биологических структур. Осмотические явления иногда используются в промышленности, например при получении некоторых полимерных материалов, очистке высоко-минерализованной воды методом «обратного» осмоса жидкостей.Клетки растений используют осмос также для увеличения объёма вакуоли, чтобы она распирала стенки клетки (тургорное давление). Клетки растений делают это путём запасания сахарозы. Увеличивая или уменьшая концентрацию сахарозы в цитоплазме, клетки могут регулировать осмос. За счёт этого повышается упругость растения в целом. С изменениями тургорного давления связаны многие движения растений (например, движения усов гороха и других лазающих растений). Пресноводные простейшие также имеют вакуоль, но задача вакуолей простейших заключается лишь в откачивании лишней воды из цитоплазмы для поддержания постоянной концентрации растворённых в ней веществ.
Осмос также играет большую роль в экологии водоёмов. Если концентрация соли и других веществ в воде поднимется или упадёт, то обитатели этих вод погибнут из-за пагубного воздействия осмоса.
Вант-Гоффа закон
Вант-Гоффа закон осмотического давления, определяет давление молекул растворённого вещества на полупроницаемую перепонку, отделяющую раствор от чистого растворителя и непроницаемую для растворённого вещества
Давление насыщенного пара является важным свойством раствора.
Если к чистому растворителю с давлением насыщенного пара р0 добавить постороннее нелетучее вещество, то давление пара растворителя над раствором изменится. При растворении какого-либо нелетучего вещества в данном растворителе понижается концентрация молекул последнего в единице объема жидкости, и уменьшается число молекул, вылетающих в единицу времени из жидкой фазы в парообразную. При меньшей концентрации пара, т.е. при меньшем его давлении, установится равновесие.
Следовательно, давление насыщенного пара растворителя над раствором всегда меньше, чем над чистым растворителем. Понижение давления насыщенного пара растворителя над раствором тем больше, чем выше концентрация растворенного вещества. Для идеальных растворов давление насыщенного пара определяется законом Рауля, установленного им в 1886 г.: относительное понижение давления насыщенного пара растворителя над раствором равно молярной доле растворенного вещества, т.е. отношению количества данного вещества к общему количеству растворителя и растворенного вещества:
Др / р0 = р0 - р/р0 = n2 / n1 + n2 = х2, где
р0 - давление пара растворителя над чистым растворителем,
р - давление пара растворителя над раствором,
Др / р0 - относительное понижение давления пара растворителя,
n2 - количество растворенного вещества,
n1 - количество вещества растворителя,
х2 - молярная доля растворенного вещества.
Это уравнение можно представить в другом виде и придать закону иную формулировку:
![]()
давление пара над раствором равно произведению давления пара над чистым растворителем на молярную долю растворителя.
Для очень разбавленных растворов уравнение имеет вид:
![]()
m1 и m2 - массы растворителя и растворенного вещества соответственно,
М1 и М2 - молярные массы растворителя и растворенного вещества соответственно.
2) Понижение давления насыщенного пара влечет за собой понижение температуры замерзания раствора по сравнению с чистым растворителем.
Жидкость замерзает при той температуре, при которой давление насыщенного пара над ней такое же, как и над кристаллами этого вещества. Так как давление насыщенного пара растворителя над раствором всегда меньше, чем над чистым растворителем, то разбавленный раствор будет замерзать при более низкой температуре, чем растворитель. Температурой замерзания раствора считают ту температуру, при которой в процессе охлаждения начинают выделяться первые кристаллы чистого растворителя. Для таких растворов Рауль нашел, что понижение температуры замерзания раствора Дtз. = t0 - t (t0 - температура замерзания растворителя, t - температура замерзания раствора) пропорционально его моляльности (1 моль в 1000 г растворителя):
Дtз = К · m, где
Дt - понижение температуры замерзания,
m - моляльность раствора,
К - криоскопическая постоянная ("криос" - холод).
Физический смысл криоскопической постоянной К состоит в том, что она равна понижению tз. раствора, содержащего 1 моль растворенного вещества на 1 кг растворителя. Величина К зависит от природы растворенного вещества, если его молекулы не диссоциируют и не ассоциируют.
Величину К можно рассчитать по формуле
![]()
T0 -температура замерзания растворителя,
l пл - удельная теплота плавления растворителя.
Так как
![]() [моль/кг], то
[моль/кг], то
![]()
m1 - масса растворителя,
m2 - масса растворенного вещества.
3) Повышение температуры кипения.
Жидкость закипает при температуре, при которой давление насыщенного пара жидкости становится равным внешнему давлению. Так как давление насыщенного пара растворов нелетучих или малолетучих веществ меньше давления насыщенного пара растворителя, то эти растворы кипят при более высокой температуре, чем растворитель. Для разбавленных растворов таких веществ Рауль установил, что повышение температуры кипения раствора Дtк. = t - t0 пропорционально его моляльности:
Дtк. = Е · m, где
Е - эбуллиоскопическая постоянная ("эбуллиос" - кипеть),
m - моляльность раствора.
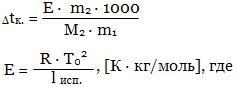
T0 -температура кипения растворителя,
lисп. - удельная теплота испарения растворителя. Величина Е численно равна повышению tк моляльного раствора при условии сохранения свойств раствора до этой концентрации. Метод определения молярной массы на основе измерения понижения температуры замерзания Дtз растворов - метод криоскопии, на основе измерения повышения температуры кипения Дtк. - эбуллиоскопический метод. Более распространен первый метод, так как Дtк растворов обычно мала и на нее влияют такие факторы как изменение внешнего давления, перегрев раствора и т.д. Одним из важных свойств растворов является осмос. Если отделить раствор от растворителя мембраной, проницаемой лишь для частиц растворителя, то будет наблюдаться самопроизвольный переход растворителя в раствор. Явление самопроизвольной диффузии растворителя через полупроницаемую перегородку в раствор называется осмосом. Сила, рассчитанная на единицу площади мембраны, заставляющая переходить растворитель через полупроницаемую перегородку в раствор, называется осмотическим давлением. По определению Вант-Гоффа осмотическое давление разбавленных растворов численно равно произведению молярной концентрации и температуры:
р = с · R · T,
где с - молярная концентрация.
Так как с = n / v, где n - количество вещества (моль), v - объем раствора (л), то
р = n / v · R · T,
Зависимость осмотического давления от понижения давления пара растворителя над раствором выражается уравнением:
![]()
где М0 - молярная масса растворителя. Уравнение Вант-Гоффа распространяется только на разбавленные растворы. При изучении свойств растворов электролитов (кислоты, щелочи, соли) было обнаружено, что этим растворам присущи все основные свойства растворов неэлектролитов, такие, как понижение давления насыщенного пара над раствором, понижение температуры замерзания, повышение температуры кипения и осмотическое давление. Однако эти величины для растворов электролитов оказались иными, чем следовало ожидать, исходя из их молярных концентраций.
Вант-Гофф, чтобы применить законы растворов неэлектролитов к электролитам, ввел поправочный множитель, названный изотоническим коэффициентом i. Этот коэффициент показывал, во сколько раз наблюдаемое осмотическое давление (рtз., рtк.) больше вычисленного, т.е.
р = i · с · R · T
Дtз = i · К · m
и т.д.
Для растворов неэлектролитов i = 1, для электролитов - i > 1.
В 1887 году С. Аррениус выдвинул гипотезу, согласно которой в растворах электролитов молекулы распадаются на ионы - катионы и анионы. Очень скоро эта гипотеза превратилась в теорию электролитической диссоциации (ЭДС). Основные положения ее таковы:
Вещества, распадающиеся на ионы в растворах или расплавах, и потому проводящие электрический ток, называются электролитами.
Электролиты при растворении в воде распадаются на ионы: положительные - катионы и отрицательные - анионы.
Диссоциация - процесс для большинства электролитов обратимый: наряду с распадом протекает процесс соединения ионов (ассоциация).
Водные растворы электролитов условно делятся на три группы: сильные, средние и слабые. Всвязи с чем введена величина степень диссоциации б = nдис. / nобщ., где nдис. - число молекул электролита, распавшихся на ионы, nобщ. - общее число молекул электролита в растворе.
Если б > 30% - это сильные электролиты, б = 5-30% имеют средние электролиты, б < 5% - слабые электролиты.
24.Растворы электролитов. Изотонический коэффициент. Теория электролитической диссоциации. Степень электролитической диссоциации. Понятие об активности
Электролитическая диссоциация, полный или частичный распад молекул растворенного вещества на катионы и анионы. Электролитической диссоциацией называют также распад на катионы и анионы ионных кристаллов при растворении или расплавлении. Электролитическая диссоциация, как правило, происходит в полярных растворителях.
При
электролитической
диссоциации
разрываются
обычно лишь
наиболее полярные
связи молекул,
например карбоновые
кислоты RCOOH диссоциируют
на ![]() и Н+, электролитической
диссоциации
могут подвергаться
молекулы некоторых
растворителей,
например воды.
и Н+, электролитической
диссоциации
могут подвергаться
молекулы некоторых
растворителей,
например воды.
Основными причинами электролитической диссоциации являются, с одной стороны, взаимодействие растворенного вещества с растворителем, которое приводит к сольватации ионов, а с другой стороны - значительное ослабление электростатических взаимодействий между сольватированными ионами в среде, обусловленное ее электростатическим полем (диэлектрической проницаемостью растворителя). При этом работа, необходимая для разрушения молекул (кристаллической решетки), обеспечивается за счет энергии сольватации. Электролитическая диссоциация лежит в основе деления растворов на два класса - растворы неэлектролитов и растворы электролитов. Наблюдаемое различие в коллигативных свойствах разбавленных растворов электролитов и неэлектролитов объясняется тем, что из-за электролитической диссоциации увеличивается общее число частиц в растворе. Это, в частности, приводит к увеличению осмотического давления раствора сравнительнос растворами неэлектролитов, понижению давления пара растворителя над раствором, увеличениюизменения температуры кипения и замерзания раствора относительно чистого растворителя. Электролитической диссоциацией объясняется также ионная электропроводность электролитов.
Мерой
электролитической
диссоциации
является степень
диссоциации
альфа- отношение
кол-ва диссоциированных
на ионы молекул
электролита
к их исходному
количеству
в растворе.
Согласно этому
определению
альфа- изменяется
от 0 (отсутствие
диссоциации)
до 1 (полная
диссоциация)
и зависит от
природы растворенного
вещества и
растворителя,
а также от
концентрации
раствора и
температуры.
Как правило,
с увеличением
диэлектрической
проницаемости
растворителя
![]() его
его ![]() увеличивается,
хотя заметная
диссоциация
наблюдается
в некоторых
растворителях
с низкой
увеличивается,
хотя заметная
диссоциация
наблюдается
в некоторых
растворителях
с низкой ![]() .
Способность
данного вещества
MX к электролитическая
диссоциация
в определенном
р-рителе по
схеме MX
.
Способность
данного вещества
MX к электролитическая
диссоциация
в определенном
р-рителе по
схеме MX ![]() M+
+ Х- характеризуется
константой
электролитической
диссоциации
KD, связанной,
согласно закону
действующих
масс, со степенью
диссоциации
альфа соотношением:
M+
+ Х- характеризуется
константой
электролитической
диссоциации
KD, связанной,
согласно закону
действующих
масс, со степенью
диссоциации
альфа соотношением:
![]()
где
х: - молярная
концентрация
электролита![]() -
средний ионный
коэффициент
активности;
-
средний ионный
коэффициент
активности;
![]() коэффициент
активности
недиссоциированной
части электролита.
Как и значение
константы KD
зависит от
свойств растворенного
вещества, в
частности от
прочности связи
между фрагментами
молекул электролита,
образующими
катион и анион,
от диэлектрических
свойств растворителя,
его способности
сольватировать
ионы, а также
от температуры
и давления; в
отличие от
альфа не зависит
от концентрации
раствора. Константа
KD может быть
определена
экспериментально,
например, по
зависимости
электропроводности
раствора от
концентрации
электролита
или путем прямого
измерения
содержания
свободных ионов
в растворе,
например,
спектрофотометрическим
методом.
коэффициент
активности
недиссоциированной
части электролита.
Как и значение
константы KD
зависит от
свойств растворенного
вещества, в
частности от
прочности связи
между фрагментами
молекул электролита,
образующими
катион и анион,
от диэлектрических
свойств растворителя,
его способности
сольватировать
ионы, а также
от температуры
и давления; в
отличие от
альфа не зависит
от концентрации
раствора. Константа
KD может быть
определена
экспериментально,
например, по
зависимости
электропроводности
раствора от
концентрации
электролита
или путем прямого
измерения
содержания
свободных ионов
в растворе,
например,
спектрофотометрическим
методом.
Соответственно
понятиям полной
и неполной
электролитической
диссоциации
электролиты
классифицируют
на сильные ![]() и слабые
и слабые ![]() (см.
Электролиты),
полностью
диссоциируют
в растворе
многие соли
неорганических
кислот, некоторые
кислоты и основания.
Неполная
электролитическая
диссоциация
наблюдается
для солей, катионы
которых склонны
к образованию
ковалентных
связей с анионами,
например соли
Ag, Cd, Zn. Некоторые
многоосновные
кислоты, например
H2SO4, полностью
диссоциируют
лишь в отношении
отщепления
одного иона
Н+, а дальнейшая
диссоциация
(см.
Электролиты),
полностью
диссоциируют
в растворе
многие соли
неорганических
кислот, некоторые
кислоты и основания.
Неполная
электролитическая
диссоциация
наблюдается
для солей, катионы
которых склонны
к образованию
ковалентных
связей с анионами,
например соли
Ag, Cd, Zn. Некоторые
многоосновные
кислоты, например
H2SO4, полностью
диссоциируют
лишь в отношении
отщепления
одного иона
Н+, а дальнейшая
диссоциация
![]() затруднена.
Разбавленные
растворы слабых
электролитов
по своим свойствам
близки к идеальным
растворам, для
них в формуле
(1) коэффициент
активности
можно считать
равными 1. Тогда
формула (1) переходит
в закон разведения
Оствальда:
затруднена.
Разбавленные
растворы слабых
электролитов
по своим свойствам
близки к идеальным
растворам, для
них в формуле
(1) коэффициент
активности
можно считать
равными 1. Тогда
формула (1) переходит
в закон разведения
Оствальда:
![]()
в
котором а можно
заменить отношением![]() где
где ![]() и
и ![]() -соответственно
эквивалентная
электропроводность
раствора при
данной концентрации
и при бесконечном
разведении.
В соответствии
с законом Оствальда
с уменьшением
концентрации
раствора степень
диссоциации
а и эквивалентная
электропроводность
возрастают,
причем при
бесконечном
разведении
-соответственно
эквивалентная
электропроводность
раствора при
данной концентрации
и при бесконечном
разведении.
В соответствии
с законом Оствальда
с уменьшением
концентрации
раствора степень
диссоциации
а и эквивалентная
электропроводность
возрастают,
причем при
бесконечном
разведении
![]() и
и ![]() Растворы сильных
электролитов
не являются
идеальными
и для их описания
необходим учет
межионного
взаимодействия
даже в области
предельного
разведения.
При определенных
условиях, например
в растворителях
с малой диэлектрической
проницаемостью,
при низких
температурах
или при образовании
многовалентных
ионов, благодаря
сильному
электростатическому
притяжению
противоположно
заряженных
ионов могут
образовываться
ионные ассоциаты,
простейшими
из которых
являются ионные
пары.
Растворы сильных
электролитов
не являются
идеальными
и для их описания
необходим учет
межионного
взаимодействия
даже в области
предельного
разведения.
При определенных
условиях, например
в растворителях
с малой диэлектрической
проницаемостью,
при низких
температурах
или при образовании
многовалентных
ионов, благодаря
сильному
электростатическому
притяжению
противоположно
заряженных
ионов могут
образовываться
ионные ассоциаты,
простейшими
из которых
являются ионные
пары.
Равновесие между сольватированными ионами и ионными парами характеризуется константой диссоциации, аналогично исходному распаду молекул, или обратной ей величиной - константой ассоциации. В приближении электростатического взаимодействия между ионами константа диссоциации контактных ионных пар, образованных двумя ионами с радиусами r+ и r. и зарядовыми числами z+ и z-, может быть рассчитана по формуле:
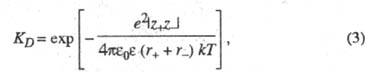
где
е - элементарный
электрический
заряд; k - постоянная
Больцмана; ![]() электрическая
постоянная
(диэлектрическая
проницаемость
вакуума) ;
электрическая
постоянная
(диэлектрическая
проницаемость
вакуума) ; ![]() -
диэлектрическая
проницаемость
растворителя;
Т - абс. температура.
-
диэлектрическая
проницаемость
растворителя;
Т - абс. температура.
Понятие электролитической диссоциации было введено С. Аррениусом в 1887. Электролитическая диссоциация играет важную роль во многих природных и производств, процессах, определяя как свойства растворов электролитов, так и особенности происходящих в них процессов.
Электролиты. Известно, что существуют две основные причины прохождения электрического тока через проводники: либо за счет движения электронов в электрическом поле, либо за счет движения ионов. Электронная проводимость присуща, прежде всего, металлам.
Ионная проводимость присуща многим химическим соединениям, обладающим ионным строением, например солям в твердом или расплавленном состояниях, а также многим водным и неводным растворам. В связи с этим все вещества принято условно делить по их поведению в растворах на две категории: а) вещества, растворы которых обладают ионной проводимостью (электролиты); б) вещества, растворы которых не обладают ионной проводимостью (неэлектролиты). К электролитам относится большинство неорганических кислот, оснований и солей. К неэлектролитам относятся многие органические соединения, например спирты, углеводы. Электролитическая диссоциация. Кроме хорошей электропроводности, растворы электролитов обладают более низкими значениями давления пара растворителя и температуры плавления и более высокими температурами кипения по сравнению с соответствующими значениями для чистого растворителя или для раствора неэлектролита в этом же растворителе. Для объяснения этих свойств шведский ученый С. Аррениус в 1887 г. предложил теорию электролитической диссоциации.Под электролитической диссоциацией понимается распад молекул электролита в растворе с образованием положительно и отрицательно заряженных ионов — катионов и анионов. Процесс диссоциации во всех случаях является обратимым, поэтому при написании уравнений реакции диссоциации необходимо применять знак обратимости «. Различные электролиты, согласно теории Аррениуса, диссоциируют на ионы в различной степени. Полнота распада зависит от природы электролита, его концентрации, природы растворителя, температуры.Степень диссоциации. Одним из важнейших понятий теории электролитической диссоциации Аррениуса является понятие о степени диссоциации.Степенью диссоциации а называется отношение числа молекул, распавшихся на ионы (n'), к общему числу растворенных молекул (п).
Из этого выражения очевидно, что а может изменяться от 0 (диссоциации нет) до 1 (полная диссоциация). Степень диссоциации часто выражают в процентах. Степень диссоциации электролита может быть определена только экспериментальным путем, например по измерению температуры замерзания раствора, по электропроводности раствора и т. д.Сильные и слабые электролиты. В зависимости от степени диссоциации различают электролиты сильные и слабые. Электролиты со степенью диссоциации больше 30% обычно называют сильными, со степенью диссоциации от 3 до 30% — средними, менее 3% — слабыми электролитами.К сильным электролитам относятся почти все соли, некоторые кислоты (НСl, HBr, HI, НNО3, НсlO4, Н2SO4(разб.)) и некоторые основания (LiОН, NaOH, КОН, Са(ОН)2, Sr(OH)2, Ва(ОН)2). К слабым электролитам относится большинство кислот (особенно органических) и оснований.Степень диссоциации как сильных, так и слабых электролитов зависит от концентрации раствора (степень диссоциации тем выше, чем более разбавлен раствор). Константа диссоциации. Более точной характеристикой диссоциации электролита является константа диссоциации, которая от концентрации раствора не зависит.Выражение для константы диссоциации можно получить, если записать уравнение реакции диссоциации электролита АК в общем виде:A K « A- + K+.
Поскольку диссоциация является обратимым равновесным процессом, то к этой реакции применим закон действующих масс, и можно определить константу равновесия как
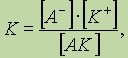
где К — константа диссоциации, которая зависит от температуры и природы электролита и растворителя, но не зависит от концентрации электролита.
Диапазон констант равновесия для разных реакций очень большой — от 10-16 до 1015. Например, высокое значение К для реакции
![]()
означает, что если в раствор, содержащий ионы серебра Ag+, внести металлическую медь, то в момент достижения равновесия концентрация ионов меди [Cu2+] намного больше, чем квадрат концентрации ионов серебра [Ag+]2. Напротив, низкое значение К в реакции
![]()
говорит о том, что к моменту достижения равновесия растворилось ничтожно малое количество иодида серебра AgI.
Обратите особое внимание на форму записи выражений для константы равновесия. Если концентрации некоторых реагентов существенно не изменяются в процессе реакции, то они не записываются в выражение для константы равновесия (такие константы обозначаются К1). Так, для реакции меди с серебром неправильным будет выражение
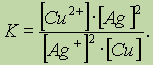
Правильной будет следующая форма записи:
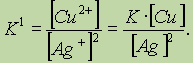
Это объясняется тем, что концентрации металлических меди и серебра введены в константу равновесия. Концентрации меди и серебра определяются их плотностью и не могут быть изменены. Поэтому эти концентрации нет смысла учитывать при расчете константы равновесия.Аналогично объясняются выражения констант равновесия при растворении AgCl и AgI.
Разбавленные растворы электролитов – солей, оснований, кислот в воде – показывают систематические отклонения от свойств идеальных растворов. Эти отклонения связаны с тем, что молекулы электролита в водном растворе распадаются на ионы, и в единице объёма раствора будет содержаться большее число частиц, чем в исходной загрузке соли, кислоты, основания. Для расчета свойств разбавленных растворов электролитов необходимо уравнения законов идеальных растворов исправить, введя в них коэффициент, учитывающий изменение числа частиц в растворе вследствие диссоциации или ассоциации растворенного вещества. Этот коэффициент обозначают i и называют изотоническим коэффициентом. Он показывает отношение числа частиц, образующихся в растворе, к числу частиц в исходной порции вещества. Для электролитов:
АВ = А+ + В-
N(1-б) Nб Nб
б=N1/N
N1 – число образовавшихся ионов или распавшихся молекул
N(1-б) – число нераспавшихся молекул
У Ni = N - Nб +Nб +Nб i= У Ni/N = 1+б
если исходные молекулы распадаются на н новых частиц, то
У Ni = N[1+б(н-1)] i= У Ni/N = 1+б(н-1)
если б=0, то i=1, если б=1, то 1<= i <= н
Для раствора, в котором молекулы растворенных веществ ассоциируют друг с другом:
nA = An
N(1-б) Nб/n
Nб/n – число ассоциированных молекул
N(1-б) – число исходных молекул
У Ni = N - Nб +Nб/n i= У Ni/N = 1+б(1/n -1)
1/n <= i <= 1
С учетом этой поправки законы разбавленных растворов электролитов запишутся:
р = i cRT
∆p/po = i n2/(n1 + i n2), ∆p/po = i n2/n1
i = роп/ррасч = (∆p/p)оп/(∆p/p)расч = ∆T3 оп /∆T3 расч = ∆Tк оп /∆Tк расч
Между активностью a2 сильного электролита в растворе (если формально не учитывать его диссоциацию на ионы) и средней активностью ионов электролита
y±
![]()
Рассмотрим несколько способов определения среднего коэффициента активности электролита y± по равновесным свойствам раствора электролитов.
Похожие работы
... подкрепляет своим одобрением неправильный или не вполне точный ответ ученика. 1.2 Совершенствование школьного химического эксперимента при проблемном обучении 1.2.1 Принципы разработки методической системы и содержания опытов по химии в системе проблемного обучения Характерной особенностью развивающего обучения является широкое использование проблемного подхода, который включает создание ...
ависимо от способа получения и места нахождения. 2. Строение внешнего электронного уровня атома калия и кальция. 1 правило Клечковского. Строение внешнего электронного уровня атома скандия. 2правило Клечковского У атома аргона остаются незанятыми все орбитали 3d-подуровня. Однако у следующих за аргоном элементов – калия и кальция – заполнение 3-го электронного слоя временно прекращается, и ...
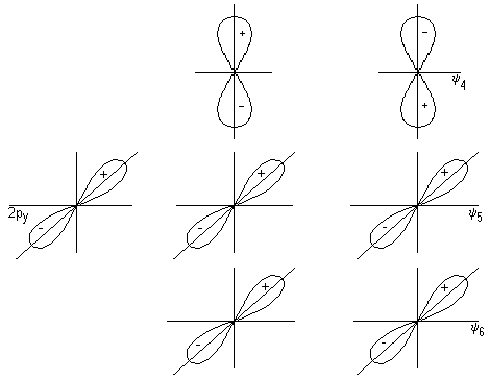
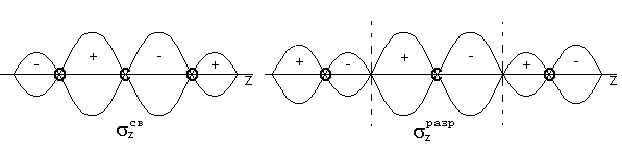
... разовая) – 0,01%. 4 Содержание Введение......................................................................................................................4 Глава 1. Межпредметные связи в курсе школьного предмета химии на примере углерода и его соединений.......................................................................5 1.1 Использование межпредметных связей для формирования у учащихся ...
... учреждение страны, а в ее задачи входило усовершенствование наук, просвещение, а также усовершенствование мануфактур, ремесел и фабрик. В то же время в начале XIX столетия, особенно после Отечественной войны 1812 г., в развитии химии в России появились новые черты. Смена мануфактурного производства фабрично-заводским выдвинула перед учеными множество практических задач, связанных с рациональной ...
0 комментариев