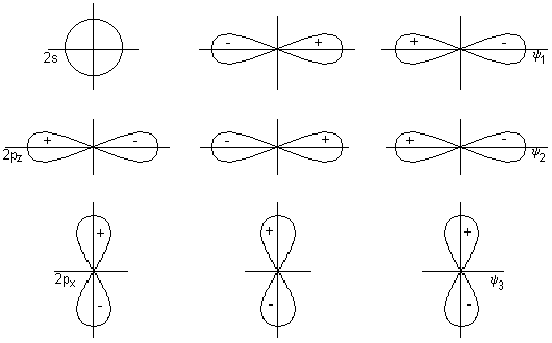
Ковалентная
(неполярная,
полярная) связь.
Механизмы
образования
ковалентной
связи
Году американский
ученый Льюис
высказал
предположение
о том, что химическая
связь образуется
за счет обобществления
двух электронов
Скорость
химических
реакций. Порядок
и молекулярность
реакций
Зависимость
скорости реакции
от температуры.
Правило Вант-Гоффа.
Уравнение
Аррениуса
Энергия
активации.
Активированные
комплексы.
Уравнение
Аррениуса
Растворы.
Физическая
и химическая
теории растворов
Фазовые
равновесия
в гетерогенных
системах, фазовые
превращения
и правило фаз.
Диаграммы
состояния
Сильные
и слабые электролиты.
Константа
диссоциации.
Закон разбавления
Оствальда.
Слабые электролиты.
Константа
диссоциации
Электролитическая
диссоциация
воды. Ионное
произведение
воды. Водородный
показатель
среды. Понятие
об индикаторах
Гидролиз
солей. Обратимый
и необратимый
(полный) гидролиз.
Роль процессов
гидролиза при
эксплуатации
котельных
установок.ъ
Растворимость
веществ. Произведение
растворимости.
Механизм
накипеообразования
Концентрации
или парциальные
давления окисленной
и восстановленной
форм
Окислитель
или восстановитель
иногда дополнительно
расходуется
на связывание
получающихся
продуктов
(солеобразование).Например,
в реакции
Электродный
потенциал.
Влияние температуры
и концентрации
на величину
электродного
потенциала.
Уравнение
Нернста
Практическое
использование
электрохимических
процессов.
Химические
источники тока
Коррозия
металлов. Основные
виды коррозии.
Химическая
коррозия
Коррозия
металлов.
Электрохимическая
коррозия
Методы защиты
металлов от
коррозии: изменение
свойств коррозионной
среды, защитные
покрытия,
электрохимическая
защита
Распространенность
химических
элементов.
Основные классы
неорганических
соединений
Навигация
Энергия активации. Активированные комплексы. Уравнение Аррениуса
Общая и неорганическая химия
442397
знаков
6
таблиц
13
изображений
17. Энергия активации. Активированные комплексы. Уравнение Аррениуса
Энергия активации, разность между значениями средней энергии частиц (молекул, радикалов, ионов и др.), вступающих в элементарный акт химической реакции, и средней энергии всех частиц, находящихся в реагирующей системе. Для различных химических реакций Э. а. изменяется в широких пределах — от нескольких до ~ 10 дж./ моль. Для одной и той же химической реакции значение Э. а. зависит от вида функций распределения молекул по энергиям их поступательного движения и внутренним степеням свободы (электронным, колебательным, вращательным). Как статистическую величину Э. а. следует отличать от пороговой энергии, или энергетического барьера, — минимальной энергии, которой должна обладать одна пара сталкивающихся частиц для протекания данной элементарной реакции.В рамках представлений теории абсолютных скоростей реакций Э. а. — разность между значениями средней энергии активированных комплексов и средней энергии исходных молекул.Представления об Э. а. возникли в 70—80-х гг. 19 в. в результате работ Я. Вант-Гоффа и С. Аррениуса, посвященных изучению влияния температуры на скорость химической реакции. Константа скорости реакции k связана с Э. а. (Е) уравнением Аррениуса: k = koe-E/RT где R — газовая постоянная, Т — абсолютная температура в К, ko — постоянная, называемая предэкспоненциальным множителем константы скорости. Это уравнение, основанное на молекулярно-кинетической теории, позже было получено в статистической физике с учетом ряда упрощающих предположений, одно из которых — независимость Э. а. от температуры. Для практики и для теоретических расчетов в сравнительно узких температурных интервалах это предположение справедливо.
Э. а. можно найти по экспериментальным данным несколькими способами. Согласно одному из них, исследуют кинетику реакции при нескольких температурах (о методах см. в ст. Скорость химической реакции) и строят график в координатах In k — 1/T; тангенс угла наклона прямой на этом графике, в соответствии с уравнением Аррениуса, равен Е. Для одностадийных обратимых реакций (см. Обратимые и необратимые реакции) Э. а. реакции в одном из направлений (прямом или обратном) можно вычислить, если известна Э. а. реакции в другом и температурная зависимость константы равновесия (из термодинамических данных). Для более точных расчетов следует учитывать зависимость Э. а. от температуры. Э. а. сложных реакций представляет собой комбинацию Э. а. элементарных стадий. Иногда, помимо истинной Э. а., определяемой по уравнению Аррениуса, используют понятие "кажущейся" Э. а. Например, если константы скоростей гетерогенно-каталитических реакций определяют по изменению объемных концентраций исходных веществ и продуктов, то кажущаяся Э. а. отличается от истинной на величину тепловых эффектов, сопровождающих процессы адсорбции и десорбции реагирующих веществ на поверхности катализатора. В неравновесных системах, например плазмохимических (см. Плазмохимия), определение Э. а. является очень сложной задачей. В некоторых случаях, однако, возможно формальное применение уравнения Аррениуса. Э. а. — важнейшее понятие кинетики химической; ее значения включают в специальные справочники и используют в химической технологии для расчета скоростей реакций в различных условиях.
В теории активированного комплекса для любой элементарной реакции предполагается, что начальная конфигурация атомов переходит в конечную в результате непрерывного изменения межъядерных расстояний.Например в ходе элементарной реакции А + ВС -> АВ + С сближаются атомы А и В. Расстояние А-В (R1) уменьшается, а расстояние В-С (R2) увеличивается.Установлено, что подобные реакции осуществляются с наименьшей затратой энергии, если атомы располагаются на одной линии, соединяющей их центры. Тогда ход реакции можно описать, использую всего два межъядерных расстояния R1 и R2. В процессе непрерывного изменения межъядерных расстояний всегда образуется промежуточная конфигурация А...В...С, в которой связь В-С уже ослаблена, но еще не полностью разорвана, а связь А-В уже начала образовываться. Такая конфигурация является критической для данной реакции. Продукты реакции могут появиться только при условии образования этой конфигурации, которая называется переходным состоянием или активированным комплексом.
18 Скорость химических реакций. Катализ. Гомогенный и гетерогенный катализ
Под скоростью химической реакции понимается изменение концентрации одного из реагирующих веществ в единицу времени. Концентрация вещества определяется числом молей в литре.При этом безразлично, о каком из участвующих в реакции веществ идет речь: псе они связаны между собой уравнениями реакции. По изменению концентрации одного из веществ можно судить о соответствующих изменениях концентраций всех остальных.При рассмотрении вопроса о скорости реакции необходимо различать гомогенные и гетерогенные реакции.Гомогенные реакции протекают в однородной среде, например в смеси газов или в водном растворе. Например:
2S02 + 02 -> 2SOsгаз газ газ
Гетерогенные реакции идут на поверхности соприкосновения твердого вещества и газа, твердого вещества и жидкости и т. д. Например:
Fe + 2HCl-> FeCl2 + H2t
твердое раствор
В связи с этим скорость гомогенной реакции и скорость гетерогенной реакции определяются различно.Скорость гомогенной реакции математически можно отразить так:
V=+,- дельта с/дельта t
v — скорость реакции в гомогенной системе;с,, с2 — начальная и конечная концентрации одного из веществ, измеряемая числом молей в литре (моль/л);г,, t2 — моменты времени (секунды, минуты).Если речь идет о концентрации исходного вещества (концентрация которого с течением времени уменьшается), то берется знак «-». Если скорость оценить увеличением концентрации одного из продуктов реакции, то берется знак «+».Скорость реакции выражается в моль/л.
Катализом
называется
явление увеличения
скорости или
возбуждения
хим. р-ии происходящей
под действием
некоторых
веществ. Вещества,
в присутствии
которых наблюдается
указанное
явление называются
катализаторами.
Катализаторы,
участвуя в
р-ии, по её завершении
вновь регенерируются,
оставаясь
неизменными.
Различают
гомогенный,
когда катализатор
и реагирующие
вещества находятся
в одной фазе,
гетерогенный,
когда катализатор
и реагенты в
разных фазах,
кроме этого
выделяют
ферментативный
катализ, когда
катализатором
являются ферменты.
Микрогетерогенный
катализ, когда
катализатор
присутствует
в каллойдном
состоянии,
размер частиц
см. особенности:
1) катализатор
изменяет скорость
лишь таких
р-ии, которые
т/д возможны
при данных
условиях ![]() .
Прежде чем
подбирать
катализатор
для той или
иной р-ии нужно
убедиться в
её т/д возможности.
.
Прежде чем
подбирать
катализатор
для той или
иной р-ии нужно
убедиться в
её т/д возможности.
2) Катализатор непосредственно участвует в р-ях, образовывая промежуточные соединения, изменяя тем самым число и вид элементарных стадий процесса. Схематично можно представить так. Пусть для р-ии А+В=С+D катализатор является вещество К, тогда в присутствии катализатора схема может быть такой А+К→АК (а), АК+В=C+D+K (б). Р-ии (а) и (б) идут во много раз быстрее, чем р-ии непосредственного взаимодействия без катализатора. Промежуточное соединение в катализе это не обычное устойчивое хим. соединение, которое может быть выделено в чистом виде или может существовать в виде отдельной фазы. В гомогенном катализе это очень не стойкие соединения с малым временем жизни. В гетерогенном катализе это поверхностное соединение, не существующее в виде отдельной фазы, свойства которого резко отличаются от свойств аналогичного соединения образовывающего объёмную фазу.
3) Катализатор не изменяет величины теплового эффекта р-ии, в противном случае имело бы место не соблюдения з. сохранения и превращения энергии.
4) Катализатор не изменяет величины константы равновесия ∆H,∆S, ∆U, ∆F. Это означает, что равновесие, выход в присутствии катализатора остаётся тем же самым. Катализатор изменяет кинетические характеристики р-ии (EАКТ и предэкспоненты PZ0). действие катализатора может быть объяснено энергией активации процесса. Е3 и Е4 – энергии активации при образовании промежуточных соединений (а) и при его распаде (б), вследствие того, что энергия активации Е1 заменяет меньшую энергию Е3 и Е4 2-ух последних стадий скорость р-ии возрастает, даже если Е3+ Е4.>Е1 CH3CHO→CH4+CO при t=518єC ЕАКТ=45.5 ккал/моль в присутствии паров J2 энергия активации снижается до ЕАКТ=32,5 ккал/моль. Скорость возрастает в 10000 раз. Катализатор ускоряет и прямую и обратную р-ии в равной мере, при этом константа равновесия не изменяется. А время достижения системой равновесного состояния уменьшается.
5) Катализатор действует избирательно. Различные катализаторы могут или одну р-ию или группу р-ий или же р-ии различного класса в соответствии с этим катализаторы могут обладать индивидуальной специфичностью, групповой специфики или являться универсальными. Особое значение катализаторов имеет и используется при протекании в параллельных р-иях.
6) В гетерогенном катализе большое влияние на процесс имеет адсорбционная способность твердых катализатора, состояние его поверхности, способов и методов обработки поверхности, присутствие на поверхности атомов других элементов и т.д.
Например: введённые в катализатор некоторые добавки, которые сами не обладают каталитической активностью, могут сильно повысить активность катализатора, такие добавки называются промоторами. Наоборот присутствие некоторых др. веществ на поверхности катализатора может сильно снизить его каталитическую активность, такие вещества называются каталитическими ядрами.
Гомогенный катализ
Среди многочисленных каталитических реакций особое место занимает катализ в цепных реакциях.
“Цепными реакциями, как известно, называются такие химические и физические процессы, в которых образование в веществе или в смеси веществ некоторых активных частиц (активных центров) приводит к тому, что каждая из активных частиц вызывает целый ряд (цепь) последовательных превращений вещества” (Эмануэль, 1957).
Такой механизм развития процесса возможен благодаря тому, что активная частица взаимодействует с веществом, образуя не только продукты реакции, но и новую активную частицу (одну, две или более), способную к новой реакции превращения вещества, и т. д. Возникающая при этом цепь превращений вещества продолжается до тех пор, пока активная частица не исчезает из системы (происходит “гибель” активной частицы и обрыв цепи). Наиболее трудная стадия при этом - зарождение активных частиц (например, свободных радикалов), после же зарождения цепь превращений осуществляется легко.
Цепные реакции широко распространены в природе. Полимеризация, хлорирование, окисление и многие другие химические процессы идут по цепному, а точнее - по радикально-цепному (с участием радикалов) механизму. Механизм окисления органических соединений (на ранних стадрях) в настоящее время установлен достаточно тщательно. Если обозначить окисляющееся вещество R-H (где Н - атом водорода, имеющий наименьшую прочность связи с остальной молекулой R), то этот механизм можно записать в следующем виде: Катализаторы, например соединения металлов переменной валентности, могут оказывать влияние на любую из рассмотренных стадий процесса. Остановимся теперь на роли катализаторов в процессах вырожденного разветвления цепей. Взаимодействие гидроперекиси с металлом может приводить как к ускорению так и к торможению реакции окисления органических веществ соединениями металлов переменной валентности в зависимости от характера продуктов, образующихся при распаде гидроперекиси. Соединения металлов образуют с гидроперекисями комплекс, который распадается в “клетке” растворителя среды, если обра-зующиеся при распаде комплекса радикалы успеют выйти из клетки, то они инициируют процесс (положительный катализ). Если же эти радикалы не успеют выйти и рекомбинируют в клетке в молекулярные неактивные продукты, то это приведет к замедлению радикально-цепного процесса (отрицательный катализ), поскольку в этом случае гидроперекись - потенциальный поставщик новых радикалов- расходуется вхолостую. До сих пор мы рассматривали лишь неглубокие стадии процессов окисления; на более глубоких стадиях например в случае окисления углеводородов, образуются кислоты, спирты, кетоны, альдегиды, которые также могут реагировать с катализатором и служить дополнительным источником свободных радикалов в реакции, т. е. в этом случае будет налицо дополнительное вырожденное разветвление цепей.
Гетерогенный катализ
К сожалению, до сих пор, несмотря на достаточно большое число теорий и гипотез в области катализа, многие основополагающие открытия были сделаны случайно или в результате простого эмпирического подхода. Как известно, случайно был найден ртутный катализатор сульфирования ароматических углеводородов М. А. Ильинским, который нечаянно разбил ртутный термометр: ртуть попала в реактор, и реакция пошла. Аналогичным образом были обнаружены теперь всем хорошо известные, а в свое время открывшие новую эру в процессе полимеризации катализаторы стереоспецифической полимеризации Циглера. Естественно, что такой путь развития учения о катализе не соответствует современному уровню науки, и именно этим объясняется повышенный интерес к изучению элементарных стадий процессов в гетерогенно-каталитических реакциях. Эти исследования - прелюдия для создания строго научных основ подбора высокоэффективных катализаторов.
Во многих случаях роль гетерогенных катализаторов в процессе окисления сводится к адсорбции органического соединения и кислорода с образованием на поверхности катализатора адсорбированного комплекса этих веществ. Такой комплекс разрыхляет связи компонентов и делает их более реакционноспособными. В некоторых случаях катализатор адсорбирует лишь один компонент, который диссоциирует на радикалы. Например, пропилен на закиси меди диссоциирует с образованием аллильного радикала, легко вступающего затем в реакцию с кислородом.
Выяснилось, что каталитическая активность металлов переменной валентности в значительной мере зависит от заполнения d-орбиталей в катионах окислов металлов.
По каталитической- активности в реакции разложения многих гидроперекисей соединения металлов располагаются следующим ря-
Мы рассмотрели один из возмжных путей инициирования процесса - взаимодействие гидроперекиси с катализатором. Однако в случае окисления реакция гетерогенного инциирования цепей может протекать как путем распада на радикалы гидроперекиси, так и путем взаимодействия углеводорода с кислородом, активированным поверхностью катализатора. Инициирование цепей может быть обусловлено участием заряженной формы органического соединения RH+, образующегося при взаимодействии RH с катализатором. Так обстоит дело с катализом в реакциях инициирования (зарождения и разветвления) цепей. Роль гетерогенных катализаторов в реакциях продолжения цепи особенно четко подчеркивается изменением скорости и направления изомеризации перекисных радикалов.
19 Химическое равновесие. Факторы, влияющие на смещение равновесия. Принцип Ле Шателье. Константа химического равновесия, ее связь с термодинамическими функциями
Химические реакции заключаются во взаимодействии реагентов с образованием продуктов. Не следует, однако полагать, что направление химической реакции только одно. В действительности, химические реакции протекают и в прямом, и в обратном направлениях. Реагенты n Продукты. Такие реакции называются обратимыми. Обратимые (и необратимые) химические реакции бывают как гомогенными, так и гетерогенными. Гомогенными называют реакции, протекающие в одной фазе – газовой или жидкой. Они характеризуются отсутствием поверхности раздела между реагентами и продуктами, взаимодействие которых осуществляется во всем объеме реакционной смеси. У гомогенных обратимых реакций все участники находятся в одном агрегатном состоянии и составляют одну фазу, например:
Реакция между газообразными веществами
СН4(г) + СО2(г) n 2СО(г) + 2Н2(г)
Реакция этирификации, протекающая в водной среде.Равновесие, имеющее место у таких реакций, называется гомогенным.Обратимые гомогенные и гетерогенные реакции в отличие от необратимых идут не конца, т.е. не до полного исчезновения реагентов: они «прекращаются» прежде, чем будут полностью израсходованы их исходные вещества (если они были взяты в стехиометрических соотношениях), поэтому в реакционной смеси у таких реакций всегда присутствуют и исходные вещества и продукты их взаимодействия. максимальный выход продуктов у них менее 100% и соответствует равновесному составу. Такие реакции протекают до установления в них определенного концентрационного предела, общего для их прямого и обратного направлений, называемого состоянием химического равновесия. Именно с наступлением последнего и связывают прекращение протекания реакции в целом. Равновесие в системе наступает в результате стремления ее к минимальному значению энергии и максимальному хаотическому распределению в пространстве, характеризующемуся энтропией. Достигнутое и неизменное при состоянии химического равновесия соотношение достаточно больших, отчетливо определяемых аналитически концентраций всех веществ называют положением химического равновесия.
Одна из важнейших количественных характеристик химического равновесия – константа равновесия, позволяющая судить о полноте протекания реакции при тех или иных условиях. В общем случае применительно к любым химическим системам, как идеальным, так и реальным, константа равновесия Кравн обратимой химической реакции есть величина постоянная при данных температуре, давлении и в данном растворителе.Для гомогенных химических равновесий, устанавливающихся в идеальных жидких и газообразных (газовых смесях) растворах, константу равновесия можно выразить через равновесные молярные концентрации и равновесные молярные доли, а для равновесий в газовых смесях – через равновесные парциальные давления: (для реакции nАА(Г) + nВВ(Г) n nD D(Г) + n F F(Г)
Кравн = [D] nD [F] n F = КС - моль/м3
[А] nА [В] n В
Кравн =Хравн DnD Хравн F n F = КХ – безразмерная величина
ХравнА nА Хравн В n В
Кравн = Рравн DnD Рравн F n F = КР – Па (атм)
РравнА nА Рравн В n В
Коэффициенты активности и фугитивности компонентов таких систем равны единице, поэтому активности становятся равными концентрациям, а фугитивности – давлениям.
Активностью (фугитивностью) называют величину, при подстановке которой вместо концентрации (парциального давления) в выражения, выведенные для идеальных систем, можно применить их к реальным системам.Константы КС, КХ, КР иногда называют концентрационными или эмпирическими константами равновесия, поскольку для их расчета используются экспериментально определяемые значения равновесной молярной концентрации (в квадратных скобках), парциальные давления компонентов рравн i, равновесные молярные доли Хравн i.
Кр = Кс (RТ)rnг ; Кр = Кх робщrnг ; Кс = Кх Vм-rnг где Vм мольный объем смеси (раствора);
rnг = (nD + n F ) – (nА + n В)
– изменение числа молей газообразных веществ, рассчитываемое по разности между суммами стехиометрических коэффициентов продуктов и реагентов. (стр 331 -333 А.А.Гуров)
Связь между константами равновесия и термодинамическими характеристиками системы устанавливается уравнениями изотермы, изобары и изохоры химической реакции. Эти уравнения впервые были выведены Вант-Гоффом (1886г), поэтому в литературе их часто называют его именем, а именно: изотерма, изобара и изохора Вант-Гоффа. Каждое из них имеет несколько разновидностей и используется либо в дифференциальной, либо в интегральной формах. Уравнение изотермы в общем виде связывает энергию Гиббса, константу равновесия и начальные (или текущие), т.е. неравновесные концентрации (активности в случае реальных растворов) или неравновесные парциальные давления (фугитивности в случае реальных газовых смесей) всех компонентов химической реакции.
(rr G)р,Т = - RТ ln Кр0 + R Т ln рDn D р F n F
рАn А р В n В
Которое и называют уравнением изотермы химической реакции в общем виде.
По уравнению изотермы вычисляют энергию Гиббса реакции, а также определяют направление и глубину ее самопроизвольного протекания при данных условиях, если известны стандартная константа равновесия для данной температуры и исходный состав произвольно приготовленной реакционной смеси, т.е. неравновесные относительные, например, парциальные давления рi всех компонентов в начальный момент времени. Очевидно, что чем больше абсолютное значение (rr G)р,Т, тем менее равновесна система.
-если в реакционной смеси присутствуют или только продукты реакции или только исходные вещества, то (rr G)р,Т ®±Ґ для любой реакции;
- (rr G)р,Тс 0 реакция протекает в обратном направлении справа налево до установления равновесия;
-(rr G)р,Тб 0 реакция протекает самопроизвольно в прямом направлении;
-(rr G)р,Т= 0 отвечает состоянию химического равновесия;
-Если величина, стоящая под знаком логарифма во втором слагаемом равна единице (это возможно, например при начальных относительных парциальных давлениях всех компонентов, равных единице, т.е. компоненты находятся в стандартных условиях), то само слагаемое равно нулю, а уравнение изотермы трансформируется:
(rr G)р,Т = - RТ ln Кр0 или rr GТ0 = - RТ ln Кр0
и его называют стандартным уравнением изотермы химической реакции или уравнением стандартного сродства. С его помощью можно вычислить Кр0 без экспериментального изучения равновесия.
Уравнение изобары химической реакции определяет зависимость константы равновесия Кр0 от температуры и в дифференциальной форме выводится на основании стандартного уравнения изотермы
rr GТ0 = - R ln Кр0
Т
и дифференциального уравнения энергии Гиббса – Гельмгольца для стандартных условий
rr GТ0 = rr НТ0 + Т d (rr GТ0)
d Т
После преобразований получают уравнение изобары:
d ln Кр0 = rr Н0Т
d Т RТ2
Анализируя его можно установить, что, чем больше абсолютная величина теплового эффекта и чем ниже температура, тем больше значение производной, следовательно, сильнее сказывается изменение температуры на константе равновесия и в большей степени смещается положение равновесия. Уравнение полезно при вычислении графическим путем тепловых эффектов реакции. Если температурная зависимость константы равновесия выражена в координатах ln Кр0 - 1/ Т.
Газофазная реакция осуществляется в закрытой системе, при постоянном объеме (V = соnst).
Уравнение изохоры химической реакции в дифференциальной форме:
d ln К с0 = rr U0Т
d Т RТ2
В интегральной форме:
ln К 0с2 = rr U0Т (Т2-Т1)
К 0с1 RТ1 Т2
Где rr U0Т - изменение стандартной внутренней энергии реакции; К с0 - безразмерная стандартная константа равновесия, выраженная через относительное равновесные молярные концентрации компонентов.
К внешним факторам, влияющим на состояние равновесия любой системы (гомогенной или гетерогенной), относятся температура, давление, внешние поля (электрическое, магнитное, гравитационное и др.). Если значение последнего фактора существенно не отличается от нормального им пренебрегают. Обычно рассматривают воздействие только двух важных факторов: температуры, давления.
Влияние изменения внешних условий на состояние равновесия устанавливается термодинамическим положением, впервые сформулированным Брауном, которое называется правилом смещения положения подвижного равновесия, или принципом Ле Шателье: если на систему, находящуюся в состоянии истинного химического равновесия, оказывать внешнее воздействие путем изменения какого-либо из условий (Сi, Т, рi. Робщ), определяющих положение равновесия, то в системе происходит изменение равновесного состава и смещение положения равновесия в направлении того процесса, протекание которого ослабляет эффект (влияние) этого воздействия
Похожие работы
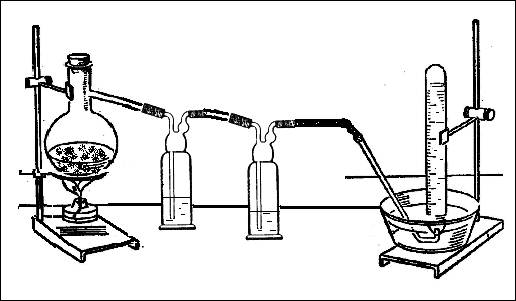
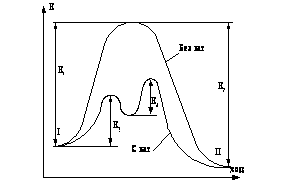
... подкрепляет своим одобрением неправильный или не вполне точный ответ ученика. 1.2 Совершенствование школьного химического эксперимента при проблемном обучении 1.2.1 Принципы разработки методической системы и содержания опытов по химии в системе проблемного обучения Характерной особенностью развивающего обучения является широкое использование проблемного подхода, который включает создание ...
ависимо от способа получения и места нахождения. 2. Строение внешнего электронного уровня атома калия и кальция. 1 правило Клечковского. Строение внешнего электронного уровня атома скандия. 2правило Клечковского У атома аргона остаются незанятыми все орбитали 3d-подуровня. Однако у следующих за аргоном элементов – калия и кальция – заполнение 3-го электронного слоя временно прекращается, и ...
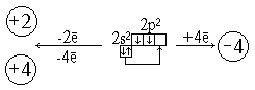
... разовая) – 0,01%. 4 Содержание Введение......................................................................................................................4 Глава 1. Межпредметные связи в курсе школьного предмета химии на примере углерода и его соединений.......................................................................5 1.1 Использование межпредметных связей для формирования у учащихся ...
... учреждение страны, а в ее задачи входило усовершенствование наук, просвещение, а также усовершенствование мануфактур, ремесел и фабрик. В то же время в начале XIX столетия, особенно после Отечественной войны 1812 г., в развитии химии в России появились новые черты. Смена мануфактурного производства фабрично-заводским выдвинула перед учеными множество практических задач, связанных с рациональной ...
0 комментариев